Zika Virus Genome and Infections
The recent outbreak of the Zika virus in Brazil and its projected spread into other countries has put the spotlight on this virus. Since this is a newly emerged virus vaccines and fast and sensitive diagnostic tools are not yet available. However, this could change fast in the near future. Specific primers for development of diagnostic tools for the detection of the Zika virus will be needed.
Zika virus is a member of the Flaviviridae family transmitted to humans by mosquitoes. It is related to other flaviviruses including dengue, West-Nile and Japanese encephalitis viruses. It produces a comparatively mild disease in humans.
See viral zone for more info: http://viralzone.expasy.org/all_by_species/43.html
Flaviviruses are small, enveloped animal viruses containing a single positive-strand genomic RNA. (Chambers et al. 1990; Flavivirus genome organization, expression, and replication.)
![]()
Since 2007 Zika virus has caused several outbreaks in the Pacific, and further spread in the Americas since 2015. These were the first documented transmissions outside of its traditional endemic areas in Africa and Asia. Zika virus is considered an emerging infectious disease with the potential to spread to new areas where the Aedes mosquito vector is present. There is no evidence of transmission Zika virus in Europe to date.
![]()
2016 Zika outbreak time line http://www.healthmap.org/zika/#timeline
Mosquito Distripution in Europe
![]()
Sources: http://ecdc.europa.eu/en/healthtopics/zika_virus_infection/Pages/index.aspx#sthash.YKl7UHeS.dpuf,
http://ecdc.europa.eu/en/healthtopics/zika_virus_infection/Pages/index.aspx
Journey of adaptation of the Plasmodium falciparum malaria parasite to New World anopheline mosquitoes plus distripution map.
http://www.scielo.br/scielo.php?script=sci_arttext&pid=S0074-02762014000500662
The genome of the Zika virus is available at PubMed:
Zika virus, complete genome
– 10,79a bp –
linear single-strand positive-strand RNA without DNA stage.
![]()
LOCUS NC_012532 10794 bp RNA linear VRL 08-FEB-2016
DEFINITION Zika virus, complete genome.
ACCESSION NC_012532
VERSION NC_012532.1 GI:226377833
DBLINK BioProject: PRJNA36615
KEYWORDS RefSeq.
SOURCE Zika virus
ORGANISM Zika virus
Viruses; ssRNA viruses; ssRNA positive-strand viruses, no DNA
stage; Flaviviridae; Flavivirus.
REFERENCE 1 (bases 1 to 10794)
AUTHORS Kuno,G. and Chang,G.-J.J.
TITLE Full-length sequencing and genomic characterization of Bagaza,
Kedougou, and Zika viruses
JOURNAL Arch Virol. 152 (4), 687-696 (2007)
PUBMED 17195954
REFERENCE 2 (bases 1 to 10794)
AUTHORS Kuno,G. and Chang,G.J.
TITLE Biological transmission of arboviruses: reexamination of and new
insights into components, mechanisms, and unique traits as well as
their evolutionary trends
JOURNAL Clin. Microbiol. Rev. 18 (4), 608-637 (2005)
PUBMED 16223950
REFERENCE 3 (bases 1 to 10794)
CONSRTM NCBI Genome Project
TITLE Direct Submission
JOURNAL Submitted (06-APR-2009) National Center for Biotechnology
Information, NIH, Bethesda, MD 20894, USA
REFERENCE 4 (bases 1 to 10794)
AUTHORS Kuno,G. and Chang,G.-J.J.
TITLE Direct Submission
JOURNAL Submitted (01-AUG-2006) Division of Vector-Borne Infect. Dis., CDC,
P.O. Box 2087, Fort Collins, CO 80522-2087, USA
REMARK Sequence update by submitter
REFERENCE 5 (bases 1 to 10794)
AUTHORS Kuno,G., Chang,G.-J.J. and Tsuchiya,K.R.
TITLE Direct Submission
JOURNAL Submitted (21-MAY-2004) Arbovirus Diseases Branch, Division of
Vector-Borne Infectious Diseases, Centers for Disease Control and
Prevention, P.O. Box 2087, Fort Collins, CO 80522, USA
COMMENT REVIEWED REFSEQ: This record has been curated by NCBI staff. The
reference sequence was derived from AY632535.
Mature peptides were annotated by RefSeq staff using the cleavage
sites reported in Kuno and Chang, 2007 (PMID 17195954). Questions
about the annotation of this sequence should be directed to
info@ncbi.nlm.nih.gov.
COMPLETENESS: full length.
FEATURES Location/Qualifiers
source 1..10794
/organism="Zika virus"
/mol_type="genomic RNA"
/strain="MR 766"
/host="sentinel monkey"
/db_xref="taxon:64320"
/country="Uganda"
/note="mosquito-borne flavivirus"
5'UTR 1..106
gene 107..10366
/gene="flavivirus polyprotein gene"
/locus_tag="ZIKV_gp1"
/db_xref="GeneID:7751225"
CDS 107..10366
/gene="flavivirus polyprotein gene"
/locus_tag="ZIKV_gp1"
/codon_start=1
/product="flavivirus polyprotein"
/protein_id="YP_002790881.1"
/db_xref="GI:226377834"
/db_xref="GeneID:7751225"
/translation="MKNPKEEIRRIRIVNMLKRGVARVNPLGGLKRLPAGLLLGHGPI
RMVLAILAFLRFTAIKPSLGLINRWGSVGKKEAMEIIKKFKKDLAAMLRIINARKERK
RRGADTSIGIIGLLLTTAMAAEITRRGSAYYMYLDRSDAGKAISFATTLGVNKCHVQI
MDLGHMCDATMSYECPMLDEGVEPDDVDCWCNTTSTWVVYGTCHHKKGEARRSRRAVT
LPSHSTRKLQTRSQTWLESREYTKHLIKVENWIFRNPGFALVAVAIAWLLGSSTSQKV
IYLVMILLIAPAYSIRCIGVSNRDFVEGMSGGTWVDVVLEHGGCVTVMAQDKPTVDIE
LVTTTVSNMAEVRSYCYEASISDMASDSRCPTQGEAYLDKQSDTQYVCKRTLVDRGWG
NGCGLFGKGSLVTCAKFTCSKKMTGKSIQPENLEYRIMLSVHGSQHSGMIGYETDEDR
AKVEVTPNSPRAEATLGGFGSLGLDCEPRTGLDFSDLYYLTMNNKHWLVHKEWFHDIP
LPWHAGADTGTPHWNNKEALVEFKDAHAKRQTVVVLGSQEGAVHTALAGALEAEMDGA
KGRLFSGHLKCRLKMDKLRLKGVSYSLCTAAFTFTKVPAETLHGTVTVEVQYAGTDGP
CKIPVQMAVDMQTLTPVGRLITANPVITESTENSKMMLELDPPFGDSYIVIGVGDKKI
THHWHRSGSTIGKAFEATVRGAKRMAVLGDTAWDFGSVGGVFNSLGKGIHQIFGAAFK
SLFGGMSWFSQILIGTLLVWLGLNTKNGSISLTCLALGGVMIFLSTAVSADVGCSVDF
SKKETRCGTGVFIYNDVEAWRDRYKYHPDSPRRLAAAVKQAWEEGICGISSVSRMENI
MWKSVEGELNAILEENGVQLTVVVGSVKNPMWRGPQRLPVPVNELPHGWKAWGKSYFV
RAAKTNNSFVVDGDTLKECPLEHRAWNSFLVEDHGFGVFHTSVWLKVREDYSLECDPA
VIGTAVKGREAAHSDLGYWIESEKNDTWRLKRAHLIEMKTCEWPKSHTLWTDGVEESD
LIIPKSLAGPLSHHNTREGYRTQVKGPWHSEELEIRFEECPGTKVYVEETCGTRGPSL
RSTTASGRVIEEWCCRECTMPPLSFRAKDGCWYGMEIRPRKEPESNLVRSMVTAGSTD
HMDHFSLGVLVILLMVQEGLKKRMTTKIIMSTSMAVLVVMILGGFSMSDLAKLVILMG
ATFAEMNTGGDVAHLALVAAFKVRPALLVSFIFRANWTPRESMLLALASCLLQTAISA
LEGDLMVLINGFALAWLAIRAMAVPRTDNIALPILAALTPLARGTLLVAWRAGLATCG
GIMLLSLKGKGSVKKNLPFVMALGLTAVRVVDPINVVGLLLLTRSGKRSWPPSEVLTA
VGLICALAGGFAKADIEMAGPMAAVGLLIVSYVVSGKSVDMYIERAGDITWEKDAEVT
GNSPRLDVALDESGDFSLVEEDGPPMREIILKVVLMAICGMNPIAIPFAAGAWYVYVK
TGKRSGALWDVPAPKEVKKGETTDGVYRVMTRRLLGSTQVGVGVMQEGVFHTMWHVTK
GAALRSGEGRLDPYWGDVKQDLVSYCGPWKLDAAWDGLSEVQLLAVPPGERARNIQTL
PGIFKTKDGDIGAVALDYPAGTSGSPILDKCGRVIGLYGNGVVIKNGSYVSAITQGKR
EEETPVECFEPSMLKKKQLTVLDLHPGAGKTRRVLPEIVREAIKKRLRTVILAPTRVV
AAEMEEALRGLPVRYMTTAVNVTHSGTEIVDLMCHATFTSRLLQPIRVPNYNLNIMDE
AHFTDPSSIAARGYISTRVEMGEAAAIFMTATPPGTRDAFPDSNSPIMDTEVEVPERA
WSSGFDWVTDHSGKTVWFVPSVRNGNEIAACLTKAGKRVIQLSRKTFETEFQKTKNQE
WDFVITTDISEMGANFKADRVIDSRRCLKPVILDGERVILAGPMPVTHASAAQRRGRI
GRNPNKPGDEYMYGGGCAETDEGHAHWLEARMLLDNIYLQDGLIASLYRPEADKVAAI
EGEFKLRTEQRKTFVELMKRGDLPVWLAYQVASAGITYTDRRWCFDGTTNNTIMEDSV
PAEVWTKYGEKRVLKPRWMDARVCSDHAALKSFKEFAAGKRGAALGVMEALGTLPGHM
TERFQEAIDNLAVLMRAETGSRPYKAAAAQLPETLETIMLLGLLGTVSLGIFFVLMRN
KGIGKMGFGMVTLGASAWLMWLSEIEPARIACVLIVVFLLLVVLIPEPEKQRSPQDNQ
MAIIIMVAVGLLGLITANELGWLERTKNDIAHLMGRREEGATMGFSMDIDLRPASAWA
IYAALTTLITPAVQHAVTTSYNNYSLMAMATQAGVLFGMGKGMPFMHGDLGVPLLMMG
CYSQLTPLTLIVAIILLVAHYMYLIPGLQAAAARAAQKRTAAGIMKNPVVDGIVVTDI
DTMTIDPQVEKKMGQVLLIAVAISSAVLLRTAWGWGEAGALITAATSTLWEGSPNKYW
NSSTATSLCNIFRGSYLAGASLIYTVTRNAGLVKRRGGGTGETLGEKWKARLNQMSAL
EFYSYKKSGITEVCREEARRALKDGVATGGHAVSRGSAKIRWLEERGYLQPYGKVVDL
GCGRGGWSYYAATIRKVQEVRGYTKGGPGHEEPMLVQSYGWNIVRLKSGVDVFHMAAE
PCDTLLCDIGESSSSPEVEETRTLRVLSMVGDWLEKRPGAFCIKVLCPYTSTMMETME
RLQRRHGGGLVRVPLCRNSTHEMYWVSGAKSNIIKSVSTTSQLLLGRMDGPRRPVKYE
EDVNLGSGTRAVASCAEAPNMKIIGRRIERIRNEHAETWFLDENHPYRTWAYHGSYEA
PTQGSASSLVNGVVRLLSKPWDVVTGVTGIAMTDTTPYGQQRVFKEKVDTRVPDPQEG
TRQVMNIVSSWLWKELGKRKRPRVCTKEEFINKVRSNAALGAIFEEEKEWKTAVEAVN
DPRFWALVDREREHHLRGECHSCVYNMMGKREKKQGEFGKAKGSRAIWYMWLGARFLE
FEALGFLNEDHWMGRENSGGGVEGLGLQRLGYILEEMNRAPGGKMYADDTAGWDTRIS
KFDLENEALITNQMEEGHRTLALAVIKYTYQNKVVKVLRPAEGGKTVMDIISRQDQRG
SGQVVTYALNTFTNLVVQLIRNMEAEEVLEMQDLWLLRKPEKVTRWLQSNGWDRLKRM
AVSGDDCVVKPIDDRFAHALRFLNDMGKVRKDTQEWKPSTGWSNWEEVPFCSHHFNKL
YLKDGRSIVVPCRHQDELIGRARVSPGAGWSIRETACLAKSYAQMWQLLYFHRRDLRL
MANAICSAVPVDWVPTGRTTWSIHGKGEWMTTEDMLMVWNRVWIEENDHMEDKTPVTK
WTDIPYLGKREDLWCGSLIGHRPRTTWAENIKDTVNMVRRIIGDEEKYMDYLSTQVRY
LGEEGSTPGVL"
mat_peptide 107..472
/gene="flavivirus polyprotein gene"
/locus_tag="ZIKV_gp1"
/product="anchored capsid protein C"
/protein_id="YP_009227206.1"
/db_xref="GI:985757037"
mat_peptide 107..418
/gene="flavivirus polyprotein gene"
/locus_tag="ZIKV_gp1"
/product="capsid protein C"
/protein_id="YP_009227196.1"
/db_xref="GI:985757027"
mat_peptide 473..976
/gene="flavivirus polyprotein gene"
/locus_tag="ZIKV_gp1"
/product="membrane glycoprotein precursor M"
/protein_id="YP_009227197.1"
/db_xref="GI:985757028"
mat_peptide 473..751
/gene="flavivirus polyprotein gene"
/locus_tag="ZIKV_gp1"
/product="protein pr"
/protein_id="YP_009227207.1"
/db_xref="GI:985757038"
mat_peptide 752..976
/gene="flavivirus polyprotein gene"
/locus_tag="ZIKV_gp1"
/product="membrane glycoprotein M"
/protein_id="YP_009227208.1"
/db_xref="GI:985757039"
mat_peptide 977..2476
/gene="flavivirus polyprotein gene"
/locus_tag="ZIKV_gp1"
/product="envelope protein E"
/protein_id="YP_009227198.1"
/db_xref="GI:985757029"
mat_peptide 2477..3532
/gene="flavivirus polyprotein gene"
/locus_tag="ZIKV_gp1"
/product="nonstructural protein NS1"
/protein_id="YP_009227199.1"
/db_xref="GI:985757030"
mat_peptide 3533..4210
/gene="flavivirus polyprotein gene"
/locus_tag="ZIKV_gp1"
/product="nonstructural protein NS2A"
/protein_id="YP_009227200.1"
/db_xref="GI:985757031"
mat_peptide 4211..4600
/gene="flavivirus polyprotein gene"
/locus_tag="ZIKV_gp1"
/product="nonstructural protein NS2B"
/protein_id="YP_009227201.1"
/db_xref="GI:985757032"
mat_peptide 4601..6451
/gene="flavivirus polyprotein gene"
/locus_tag="ZIKV_gp1"
/product="nonstructural protein NS3"
/protein_id="YP_009227202.1"
/db_xref="GI:985757033"
mat_peptide 6452..6832
/gene="flavivirus polyprotein gene"
/locus_tag="ZIKV_gp1"
/product="nonstructural protein NS4A"
/protein_id="YP_009227203.1"
/db_xref="GI:985757034"
mat_peptide 6833..6901
/gene="flavivirus polyprotein gene"
/locus_tag="ZIKV_gp1"
/product="protein 2K"
/protein_id="YP_009227209.1"
/db_xref="GI:985757040"
mat_peptide 6902..7654
/gene="flavivirus polyprotein gene"
/locus_tag="ZIKV_gp1"
/product="nonstructural protein NS4B"
/protein_id="YP_009227204.1"
/db_xref="GI:985757035"
mat_peptide 7655..10363
/gene="flavivirus polyprotein gene"
/locus_tag="ZIKV_gp1"
/product="RNA-dependent RNA polymerase NS5"
/protein_id="YP_009227205.1"
/db_xref="GI:985757036"
3'UTR 10367..10794
ORIGIN
1 agttgttgat ctgtgtgagt cagactgcga cagttcgagt ctgaagcgag agctaacaac
61 agtatcaaca ggtttaattt ggatttggaa acgagagtttctggtcatga aaaaccccaa
121 agaagaaatc cggaggatcc ggattgtcaa tatgctaaaa cgcggagtag cccgtgtaaa
181 ccccttggga ggtttgaaga ggttgccagc cggacttctg ctgggtcatg gacccatcag
241 aatggttttg gcgatactag cctttttgag atttacagca atcaagccat cactgggcct
301 tatcaacaga tggggttccg tggggaaaaa agaggctatg gaaataataa agaagttcaa
361 gaaagatctt gctgccatgt tgagaataat caatgctagg aaagagagga agagacgtgg
421 cgcagacacc agcatcggaa tcattggcct cctgctgact acagccatgg cagcagagat
481 cactagacgc gggagtgcat actacatgta cttggatagg agcgatgccg ggaaggccat
541 ttcgtttgct accacattgg gagtgaacaa gtgccacgta cagatcatgg acctcgggca
601 catgtgtgac gccaccatga gttatgagtg ccctatgctg gatgagggag tggaaccaga
661 tgatgtcgat tgctggtgca acacgacatc aacttgggtt gtgtacggaa cctgtcatca
721 caaaaaaggt gaggcacggc gatctagaag agccgtgacg ctcccttctc actctacaag
781 gaagttgcaa acgcggtcgc agacctggtt agaatcaaga gaatacacga agcacttgat
841 caaggttgaa aactggatat tcaggaaccc cgggtttgcg ctagtggccg ttgccattgc
901 ctggcttttg ggaagctcga cgagccaaaa agtcatatac ttggtcatga tactgctgat
961 tgccccggca tacagtatca ggtgcattgg agtcagcaat agagacttcg tggagggcat
1021 gtcaggtggg acctgggttg atgttgtctt ggaacatgga ggctgcgtta ccgtgatggc
1081 acaggacaag ccaacagtcg acatagagtt ggtcacgacg acggttagta acatggccga
1141 ggtaagatcc tattgctacg aggcatcgat atcggacatg gcttcggaca gtcgttgccc
1201 aacacaaggt gaagcctacc ttgacaagca atcagacact caatatgtct gcaaaagaac
1261 attagtggac agaggttggg gaaacggttg tggacttttt ggcaaaggga gcttggtgac
1321 atgtgccaag tttacgtgtt ctaagaagat gaccgggaag agcattcaac cggaaaatct
1381 ggagtatcgg ataatgctat cagtgcatgg ctcccagcat agcgggatga ttggatatga
1441 aactgacgaa gatagagcga aagtcgaggt tacgcctaat tcaccaagag cggaagcaac
1501 cttgggaggc tttggaagct taggacttga ctgtgaacca aggacaggcc ttgacttttc
1561 agatctgtat tacctgacca tgaacaataa gcattggttg gtgcacaaag agtggtttca
1621 tgacatccca ttgccttggc atgctggggc agacaccgga actccacact ggaacaacaa
1681 agaggcattg gtagaattca aggatgccca cgccaagagg caaaccgtcg tcgttctggg
1741 gagccaggaa ggagccgttc acacggctct cgctggagct ctagaggctg agatggatgg
1801 tgcaaaggga aggctgttct ctggccattt gaaatgccgc ctaaaaatgg acaagcttag
1861 attgaagggc gtgtcatatt ccttgtgcac tgcggcattc acattcacca aggtcccagc
1921 tgaaacactg catggaacag tcacagtgga ggtgcagtat gcagggacag atggaccctg
1981 caagatccca gtccagatgg cggtggacat gcagaccctg accccagttg gaaggctgat
2041 aaccgccaac cccgtgatta ctgaaagcac tgagaactca aagatgatgt tggagcttga
2101 cccaccattt ggggattctt acattgtcat aggagttggg gacaagaaaa tcacccacca
2161 ctggcatagg agtggtagca ccatcggaaa ggcatttgag gccactgtga gaggcgccaa
2221 gagaatggca gtcctggggg atacagcctg ggacttcgga tcagtcgggg gtgtgttcaa
2281 ctcactgggt aagggcattc accagatttt tggagcagcc ttcaaatcac tgtttggagg
2341 aatgtcctgg ttctcacaga tcctcatagg cacgctgcta gtgtggttag gtttgaacac
2401 aaagaatgga tctatctccc tcacatgctt ggccctgggg ggagtgatga tcttcctctc
2461 cacggctgtt tctgctgacg tggggtgctc agtggacttc tcaaaaaagg aaacgagatg
2521 tggcacgggg gtattcatct ataatgatgt tgaagcctgg agggaccggt acaagtacca
2581 tcctgactcc ccccgcagat tggcagcagc agtcaagcag gcctgggaag aggggatctg
2641 tgggatctca tccgtttcaa gaatggaaaa catcatgtgg aaatcagtag aaggggagct
2701 caatgctatc ctagaggaga atggagttca actgacagtt gttgtgggat ctgtaaaaaa
2761 ccccatgtgg agaggtccac aaagattgcc agtgcctgtg aatgagctgc cccatggctg
2821 gaaagcctgg gggaaatcgt attttgttag ggcggcaaag accaacaaca gttttgttgt
2881 cgacggtgac acactgaagg aatgtccgct tgagcacaga gcatggaata gttttcttgt
2941 ggaggatcac gggtttggag tcttccacac cagtgtctgg cttaaggtca gagaagatta
3001 ctcattagaa tgtgacccag ccgtcatagg aacagctgtt aagggaaggg aggccgcgca
3061 cagtgatctg ggctattgga ttgaaagtga aaagaatgac acatggaggc tgaagagggc
3121 ccacctgatt gagatgaaaa catgtgaatg gccaaagtct cacacattgt ggacagatgg
3181 agtagaagaa agtgatctta tcatacccaa gtctttagct ggtccactca gccaccacaa
3241 caccagagag ggttacagaa cccaagtgaa agggccatgg cacagtgaag agcttgaaat
3301 ccggtttgag gaatgtccag gcaccaaggt ttacgtggag gagacatgcg gaactagagg
3361 accatctctg agatcaacta ctgcaagtgg aagggtcatt gaggaatggt gctgtaggga
3421 atgcacaatg cccccactat cgtttcgagc aaaagacggc tgctggtatg gaatggagat
3481 aaggcccagg aaagaaccag agagcaactt agtgaggtca atggtgacag cggggtcaac
3541 cgatcatatg gaccacttct ctcttggagt gcttgtgatt ctactcatgg tgcaggaggg
3601 gttgaagaag agaatgacca caaagatcat catgagcaca tcaatggcag tgctggtagt
3661 catgatcttg ggaggatttt caatgagtga cctggccaag cttgtgatcc tgatgggtgc
3721 tactttcgca gaaatgaaca ctggaggaga tgtagctcac ttggcattgg tagcggcatt
3781 taaagtcaga ccagccttgc tggtctcctt cattttcaga gccaattgga caccccgtga
3841 gagcatgctg ctagccctgg cttcgtgtct tctgcaaact gcgatctctg ctcttgaagg
3901 tgacttgatg gtcctcatta atggatttgc tttggcctgg ttggcaattc gagcaatggc
3961 cgtgccacgc actgacaaca tcgctctacc aatcttggct gctctaacac cactagctcg
4021 aggcacactg ctcgtggcat ggagagcggg cctggctact tgtggaggga tcatgctcct
4081 ctccctgaaa gggaaaggta gtgtgaagaa gaacctgcca tttgtcatgg ccctgggatt
4141 gacagctgtg agggtagtag accctattaa tgtggtagga ctactgttac tcacaaggag
4201 tgggaagcgg agctggcccc ctagtgaagt tctcacagcc gttggcctga tatgtgcact
4261 ggccggaggg tttgccaagg cagacattga gatggctgga cccatggctg cagtaggctt
4321 gctaattgtc agctatgtgg tctcgggaaa gagtgtggac atgtacattg aaagagcagg
4381 tgacatcaca tgggaaaagg acgcggaagt cactggaaac agtcctcggc ttgacgtggc
4441 actggatgag agtggtgact tctccttggt agaggaagat ggtccaccca tgagagagat
4501 catactcaag gtggtcctga tggccatctg tggcatgaac ccaatagcta taccttttgc
4561 tgcaggagcg tggtatgtgt atgtgaagac tgggaaaagg agtggcgccc tctgggacgt
4621 gcctgctccc aaagaagtga agaaaggaga gaccacagat ggagtgtaca gagtgatgac
4681 tcgcagactg ctaggttcaa cacaggttgg agtgggagtc atgcaagagg gagtcttcca
4741 caccatgtgg cacgttacaa aaggagccgc actgaggagc ggtgagggaa gacttgatcc
4801 atactggggg gatgtcaagc aggacttggt gtcatactgt gggccttgga agttggatgc
4861 agcttgggat ggactcagcg aggtacagct tttggccgta cctcccggag agagggccag
4921 aaacattcag accctgcctg gaatattcaa gacaaaggac ggggacatcg gagcagttgc
4981 tctggactac cctgcaggga cctcaggatc tccgatccta gacaaatgtg gaagagtgat
5041 aggactctat ggcaatgggg ttgtgatcaa gaatggaagc tatgttagtg ctataaccca
5101 gggaaagagg gaggaggaga ctccggttga atgtttcgaa ccctcgatgc tgaagaagaa
5161 gcagctaact gtcttggatc tgcatccagg agccggaaaa accaggagag ttcttcctga
5221 aatagtccgt gaagccataa aaaagagact ccggacagtg atcttggcac caactagggt
5281 tgtcgctgct gagatggagg aggccttgag aggacttccg gtgcgttaca tgacaacagc
5341 agtcaacgtc acccattctg ggacagaaat cgttgatttg atgtgccatg ccactttcac
5401 ttcacgctta ctacaaccca tcagagtccc taattacaat ctcaacatca tggatgaagc
5461 ccacttcaca gacccctcaa gtatagctgc aagaggatac atatcaacaa gggttgaaat
5521 gggcgaggcg gctgccattt ttatgactgc cacaccacca ggaacccgtg atgcgtttcc
5581 tgactctaac tcaccaatca tggacacaga agtggaagtc ccagagagag cctggagctc
5641 aggctttgat tgggtgacag accattctgg gaaaacagtt tggttcgttc caagcgtgag
5701 aaacggaaat gaaatcgcag cctgtctgac aaaggctgga aagcgggtca tacagctcag
5761 caggaagact tttgagacag aatttcagaa aacaaaaaat caagagtggg actttgtcat
5821 aacaactgac atctcagaga tgggcgccaa cttcaaggct gaccgggtca tagactctag
5881 gagatgccta aaaccagtca tacttgatgg tgagagagtc atcttggctg ggcccatgcc
5941 tgtcacgcat gctagtgctg ctcagaggag aggacgtata ggcaggaacc ctaacaaacc
6001 tggagatgag tacatgtatg gaggtgggtg tgcagagact gatgaaggcc atgcacactg
6061 gcttgaagca agaatgcttc ttgacaacat ctacctccag gatggcctca tagcctcgct
6121 ctatcggcct gaggccgata aggtagccgc cattgaggga gagtttaagc tgaggacaga
6181 gcaaaggaag accttcgtgg aactcatgaa gagaggagac cttcccgtct ggctagccta
6241 tcaggttgca tctgccggaa taacttacac agacagaaga tggtgctttg atggcacaac
6301 caacaacacc ataatggaag acagtgtacc agcagaggtt tggacaaagt atggagagaa
6361 gagagtgctc aaaccgagat ggatggatgc tagggtctgt tcagaccatg cggccctgaa
6421 gtcgttcaaa gaattcgccg ctggaaaaag aggagcggct ttgggagtaa tggaggccct
6481 gggaacactg ccaggacaca tgacagagag gtttcaggaa gccattgaca acctcgccgt
6541 gctcatgcga gcagagactg gaagcaggcc ttataaggca gcggcagccc aactgccgga
6601 gaccctagag accattatgc tcttaggttt gctgggaaca gtttcactgg ggatcttctt
6661 cgtcttgatg cggaataagg gcatcgggaa gatgggcttt ggaatggtaa cccttggggc
6721 cagtgcatgg ctcatgtggc tttcggaaat tgaaccagcc agaattgcat gtgtcctcat
6781 tgttgtgttt ttattactgg tggtgctcat acccgagcca gagaagcaaa gatctcccca
6841 agataaccag atggcaatta tcatcatggt ggcagtgggc cttctaggtt tgataactgc
6901 aaacgaactt ggatggctgg aaagaacaaa aaatgacata gctcatctaa tgggaaggag
6961 agaagaagga gcaaccatgg gattctcaat ggacattgat ctgcggccag cctccgcctg
7021 ggctatctat gccgcattga caactctcat caccccagct gtccaacatg cggtaaccac
7081 ttcatacaac aactactcct taatggcgat ggccacacaa gctggagtgc tgtttggcat
7141 gggcaaaggg atgccattta tgcatgggga ccttggagtc ccgctgctaa tgatgggttg
7201 ctattcacaa ttaacacccc tgactctgat agtagctatc attctgcttg tggcgcacta
7261 catgtacttg atcccaggcc tacaagcggc agcagcgcgt gctgcccaga aaaggacagc
7321 agctggcatc atgaagaatc ccgttgtgga tggaatagtg gtaactgaca ttgacacaat
7381 gacaatagac ccccaggtgg agaagaagat gggacaagtg ttactcatag cagtagccat
7441 ctccagtgct gtgctgctgc ggaccgcctg gggatggggg gaggctggag ctctgatcac
7501 agcagcgacc tccaccttgt gggaaggctc tccaaacaaa tactggaact cctctacagc
7561 cacctcactg tgcaacatct tcagaggaag ctatctggca ggagcttccc ttatctatac
7621 agtgacgaga aacgctggcc tggttaagag acgtggaggt gggacgggag agactctggg
7681 agagaagtgg aaagctcgtc tgaatcagat gtcggccctg gagttctact cttataaaaa
7741 gtcaggtatc actgaagtgt gtagagagga ggctcgccgt gccctcaagg atggagtggc
7801 cacaggagga catgccgtat cccggggaag tgcaaagatc agatggttgg aggagagagg
7861 atatctgcag ccctatggga aggttgttga cctcggatgt ggcagagggg gctggagcta
7921 ttatgccgcc accatccgca aagtgcagga ggtgagagga tacacaaagg gaggtcccgg
7981 tcatgaagaa cccatgctgg tgcaaagcta tgggtggaac atagttcgtc tcaagagtgg
8041 agtggacgtc ttccacatgg cggctgagcc gtgtgacact ctgctgtgtg acataggtga
8101 gtcatcatct agtcctgaag tggaagagac acgaacactc agagtgctct ctatggtggg
8161 ggactggctt gaaaaaagac caggggcctt ctgtataaag gtgctgtgcc catacaccag
8221 cactatgatg gaaaccatgg agcgactgca acgtaggcat gggggaggat tagtcagagt
8281 gccattgtgt cgcaactcca cacatgagat gtactgggtc tctggggcaa agagcaacat
8341 cataaaaagt gtgtccacca caagtcagct cctcctggga cgcatggatg gccccaggag
8401 gccagtgaaa tatgaggagg atgtgaacct cggctcgggt acacgagctg tggcaagctg
8461 tgctgaggct cctaacatga aaatcatcgg caggcgcatt gagagaatcc gcaatgaaca
8521 tgcagaaaca tggtttcttg atgaaaacca cccatacagg acatgggcct accatgggag
8581 ctacgaagcc cccacgcaag gatcagcgtc ttccctcgtg aacggggttg ttagactcct
8641 gtcaaagcct tgggacgtgg tgactggagt tacaggaata gccatgactg acaccacacc
8701 atacggccaa caaagagtct tcaaagaaaa agtggacacc agggtgccag atccccaaga
8761 aggcactcgc caggtaatga acatagtctc ttcctggctg tggaaggagc tggggaaacg
8821 caagcggcca cgcgtctgca ccaaagaaga gtttatcaac aaggtgcgca gcaatgcagc
8881 actgggagca atatttgaag aggaaaaaga atggaagacg gctgtggaag ctgtgaatga
8941 tccaaggttt tgggccctag tggataggga gagagaacac cacctgagag gagagtgtca
9001 cagctgtgtg tacaacatga tgggaaaaag agaaaagaag caaggagagt tcgggaaagc
9061 aaaaggtagc cgcgccatct ggtacatgtg gttgggagcc agattcttgg agtttgaagc
9121 ccttggattc ttgaacgagg accattggat gggaagagaa aactcaggag gtggagtcga
9181 agggttagga ttgcaaagac ttggatacat tctagaagaa atgaatcggg caccaggagg
9241 aaagatgtac gcagatgaca ctgctggctg ggacacccgc attagtaagt ttgatctgga
9301 gaatgaagct ctgattacca accaaatgga ggaagggcac agaactctgg cgttggccgt
9361 gattaaatac acataccaaa acaaagtggt gaaggttctc agaccagctg aaggaggaaa
9421 aacagttatg gacatcattt caagacaaga ccagagaggg agtggacaag ttgtcactta
9481 tgctctcaac acattcacca acttggtggt gcagcttatc cggaacatgg aagctgagga
9541 agtgttagag atgcaagact tatggttgtt gaggaagcca gagaaagtga ccagatggtt
9601 gcagagcaat ggatgggata gactcaaacg aatggcggtc agtggagatg actgcgttgt
9661 gaagccaatc gatgataggt ttgcacatgc cctcaggttc ttgaatgaca tgggaaaagt
9721 taggaaagac acacaggagt ggaaaccctc gactggatgg agcaattggg aagaagtccc
9781 gttctgctcc caccacttca acaagctgta cctcaaggat gggagatcca ttgtggtccc
9841 ttgccgccac caagatgaac tgattggccg agctcgcgtc tcaccagggg caggatggag
9901 catccgggag actgcctgtc ttgcaaaatc atatgcgcag atgtggcagc tcctttattt
9961 ccacagaaga gaccttcgac tgatggctaa tgccatttgc tcggctgtgc cagttgactg
10021 ggtaccaact gggagaacca cctggtcaat ccatggaaag ggagaatgga tgaccactga
10081 ggacatgctc atggtgtgga atagagtgtg gattgaggag aacgaccata tggaggacaa
10141 gactcctgta acaaaatgga cagacattcc ctatctagga aaaagggagg acttatggtg
10201 tggatccctt atagggcaca gaccccgcac cacttgggct gaaaacatca aagacacagt
10261 caacatggtg cgcaggatca taggtgatga agaaaagtac atggactatc tatccaccca
10321 agtccgctac ttgggtgagg aagggtccac acccggagtg ttgtaagcac caattttagt
10381 gttgtcaggc ctgctagtca gccacagttt ggggaaagct gtgcagcctg taaccccccc
10441 aggagaagct gggaaaccaa gctcatagtc aggccgagaa cgccatggca cggaagaagc
10501 catgctgcct gtgagcccct cagaggacac tgagtcaaaa aaccccacgc gcttggaagc
10561 gcaggatggg aaaagaaggt ggcgaccttc cccacccttc aatctggggc ctgaactgga
10621 gactagctgt gaatctccag cagagggact agtggttaga ggagaccccc cggaaaacgc
10681 aaaacagcat attgacgtgg gaaagaccag agactccatg agtttccacc acgctggccg
10741 ccaggcacag atcgccgaac ttcggcggcc ggtgtgggga aatccatggt ttct
//
-.-



 Figure 2: The CRISPR locus. The CRISPR locus encodes the pre-crRNA that consists of repeat and spacer sequences. Some of the repeat sequences fold into stem-loop structures. Spacer sequences are derived from invader DNA from previous cell attacks. CRISPR locus transcription starts from the leader region (black arrow) generating pre-crRNA. The pre-crRNA is subsequently processed into crRNAs, and each crRNA is specific for one invader. (Maier L-K, Fischer S, Stoll B, et al. The immune system of halophilic archaea. Mobile Genetic Elements. 2012;2(5):228-232. doi:10.4161/mge.22530).
Figure 2: The CRISPR locus. The CRISPR locus encodes the pre-crRNA that consists of repeat and spacer sequences. Some of the repeat sequences fold into stem-loop structures. Spacer sequences are derived from invader DNA from previous cell attacks. CRISPR locus transcription starts from the leader region (black arrow) generating pre-crRNA. The pre-crRNA is subsequently processed into crRNAs, and each crRNA is specific for one invader. (Maier L-K, Fischer S, Stoll B, et al. The immune system of halophilic archaea. Mobile Genetic Elements. 2012;2(5):228-232. doi:10.4161/mge.22530).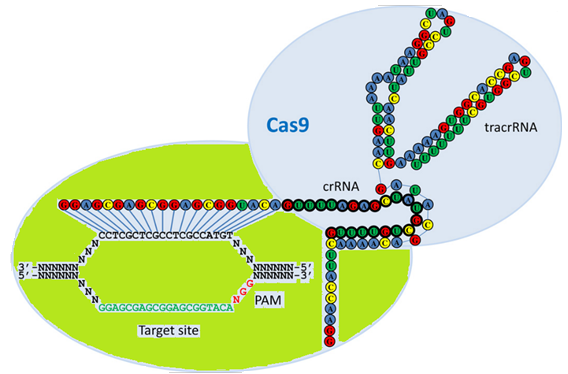
.jpg)
.png)
.png)












.jpg)
.jpg)