The Maillard reaction
The Maillard reaction is a complex set of chemical reactions between amines and carbonyl compounds such as sugars to ultimately form Amadori products.
The following scheme shows a simplistic view of the Maillard reaction:
Aldose + amino compounds -> N-substituted glycosylamines -> Amadori and fission products.
However, the complex Maillard reaction involves multiple reaction steps as will be discussed below.
Major parts of the chemistry of the Maillard reaction have been unraveled in the last decades and much of the complex reactions of the Maillard reaction is now known. However, to understand the impact of Maillard reaction products (MRPs) in human health and disease more research will need to be conducted. Close to 25 MRPs have already bene observed in body tissues and have been isolated and structurally characterized.
The Maillard reaction is a ‘non-enzymatic browning” reaction involving reduced sugars with compounds possessing free amino groups. A reactive sugar, such as glucose, can react with amino groups in amino acids, peptides, and proteins as well as with other molecules that contain free amino groups. In 1912, the French scientists Louis-Camille Maillard described the reaction between amino acids and reducing sugars during heating. The reaction generated a discolored (browning) reaction mixture. The multitude of complex reactions between amino acids and reducing sugars is now known as the Maillard reaction.
The Maillard reaction became recognized as part of the browning reactions taking place in food and beverages. A complex Maillard reaction is known to occur in virtually all heat processed and stored foods, in papers, textiles, in biopharmaceutical formulations, in the soil, as well as in glycation reactions in the mammalian body, including in the aging human body. The reaction of glucose or its autoxidation products with amines, amino acids, peptide and proteins in the human body is considered to be the first step of this complex glycation reaction leading to the formation of sugar-derived protein adducts and crosslinks in later stages. The resulting products are known as advanced glycation end-products (AGEs) observed in pathogenic stages of chronic diseases such as diabetes.
Analysis of advanced glycation end-products (AGEs)
AGE-modified proteins are usually detected and analyzed using traditional approaches such as high-performance liquid chromatography (HPLC) and capillary electrophoresis (CE), and more recently with sensitive mass spectrometry-based methods. Early studies of sugars and their derivatives by electron impact mass spectrometry were limited to volatile derivatives such as trimethylsilyl ethers, acetal derivatives, as well as acylated and methylated derivatives. However, with the development of so-called “soft” ionization techniques such as chemical ionization, field-desorption, fast-atom bombardment, and more recently electro-spray ionization and laser-desorption time-of-flight mass spectrometry, it became possible to study unmodified sugars as well as complex oligosaccharides, carbohydrates, nucleotides, peptides, proteins and related modified molecules. As a result, it is now possible to study glycoproteins as well as their oxidation products as found in AGEs.
The mutarotation reaction of sugars is the key for the initial reaction step of the Maillard reaction.
Mutarotation of glucose in aqueous solution
Freshly prepared solutions of glucose in water gradually change in optical rotary power. The cause is a mutarotation reaction in which the dissolved glucose undergoes a transformation from one form to another. Figure 1 shows the mutarotation between the a-anomer and the b-anomer of glucose.
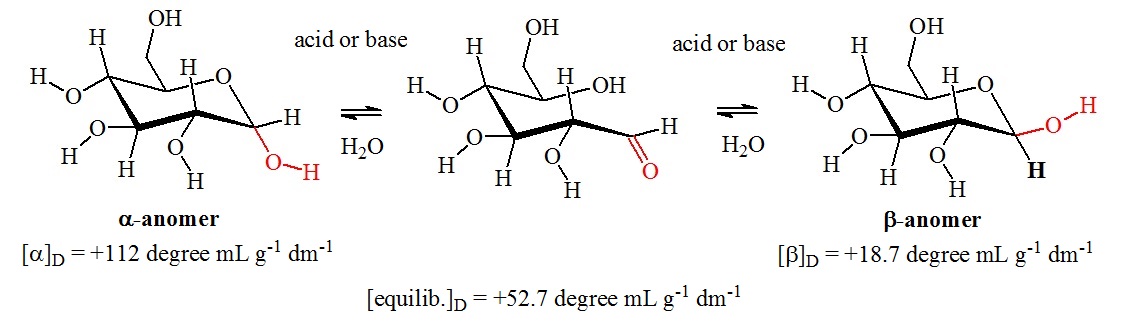
Figure 1: Mutarotation of glucose. Mutarotation is a characteristic of the cyclic hemiacetal forms of glucose. Aldehydes cannot undergo mutarotations. Mutarotation occurs by the opening of the pyranose ring to the free aldehyde form. This reaction is a reversal of a hemiacetal formation reaction. A rotation of 180° of the carbon-carbon bond to the carbonyl group allows reclosure of the hemiacetal ring via the reaction of the hydroxy group at the opposite site of the carbonyl carbon. In the glucose molecule, the two pyranose forms are interconverted. However, other carbohydrates can undergo more complex mutarotations. For example, D-fructose can mutarotate into pyranose and furanose forms.
Amadori Product Formation
Schiff base formation and Amadori rearrangement
Primary amines can react with aldehydes or ketones to form imines. This reaction is known as Schiff base formation.
Schiff base forming reaction:
R3-NH2 + R1HCO (or R1R2CO) -> R1HC=N-R3 (or R1R2C=N-R3)
The Amadori rearrangement occurs during cross-linking reactions often observed in collagen and protein glycosylation reactions. Chemically, the Amadori rearrangement refers to the conversion of N-glycosides of aldoses to N-glycosides of the corresponding ketoses. The reaction is catalyzed by acids or bases.
Steps of the Maillard reaction according to the Hodge Diagram.
1. Initial reaction between a reducing sugar and an amino group forms
an unstable Schiff base.
2. The Schiff base slowly rearranges to form Amadori products.
3. Amadori products degrade.
4. Formation of reactive carbonyl and dicarbonyl compounds.
5. Production of Strecker aldehydes from amino acids and aminoketones.
6. Production of aldol condensation products of furfurals, reductions, and
aldehydes produced during steps 3, 4 and 5.
7. Melanoidin formation: Furfurals, reductones, and aldehydes produced in
steps 3, 4, and 5 react with amino compounds to form melanoidines.
8. Free radicals can mediate the formation of carbonyl fission products
resulting from reducing sugars.
Chemistry of the Maillard Reaction and Formation of Amadori Product
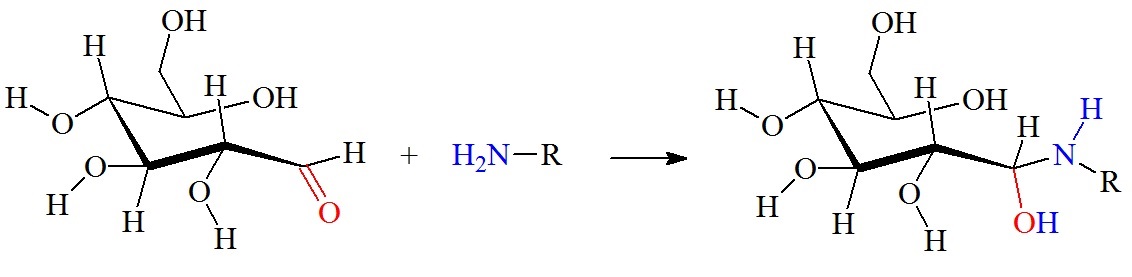
Reaction between an aldehyde group on a glucose molecule and a free amino group.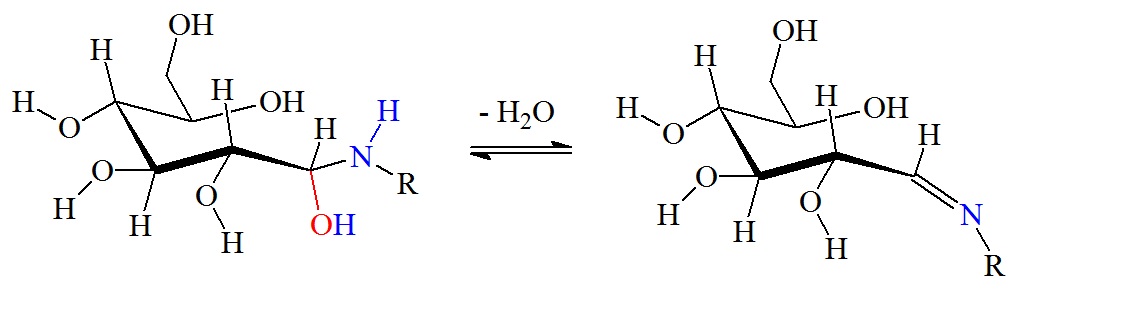
Dehydration reaction to form a Schiff base via β-elimination.
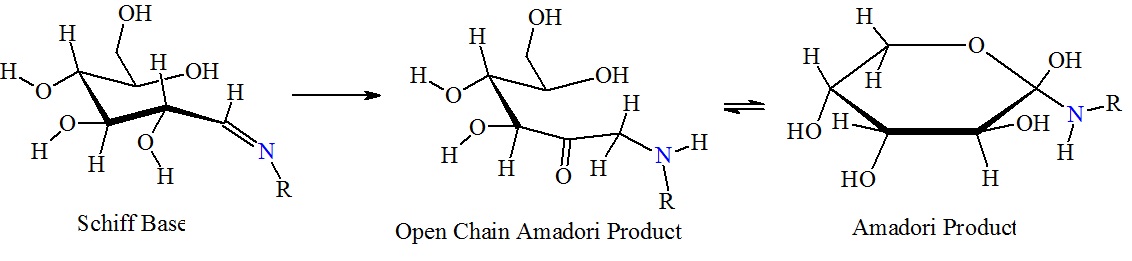
Formation of Amadori products.
Figure 2: Reaction between glucose and the amino group of amino acids, proteins or peptides. The nucleophilic attack by a free amino group on the aldehyde of glucose initially forms a carbinolamine. The carbinolamine subsequently dehydrates to a Schiff base. Next, the Schiff base undergoes a slow rearrangement to form the Amadori product. Only one Amadori product is shown here, however, due to the complexity of the Maillard reactions a mixture of several isoforms of Amadori products are generated during any Maillard reaction. Next, oxidative decomposition of Amadori products can lead to the formation of a wide range of reactive carbonyl and dicarbonyl compounds.
The Schiff base, or imine, formation is catalyzed by acids, and the dehydration of the carbiolamine is the rate-limiting step of imine formation. Imine formation is a sequence of two reactions, namely, carbonyl addition followed by β-elimination.
According to Hodge et al., browned flavors generated by the Maillard reaction are essential for the recognition and taste of many processed foods.
Browned flavors include:
(1) Food aromas that are described as toasted, baked, nutty, or roasted.
(2) Corny and amine-like aromas from cooked grains and meals. This includes
desirable and undesirable burnt aromas, bitter tastes, roasted malt, nuts,
coffee, chicory, cocoa, meats, fruits, and vegetables.
Flavor compounds isolated from browning reactions allowed correlation of aromas to chemical structures. It was found that many of these flavor compounds were formed through sugar-amines condensations followed by Amadori rearrangement at lower temperatures. The Amadori compounds 1-amino-1-deoxy-2-ketoses are important nonvolatile precursor molecules originating from Maillard reactions.

Figure 3: Maillard reaction and flvor formation in foods.
Maillard reaction products can have positive and negative effects on health. Maillard reaction products can act as antioxidants, bactericidal compounds, as antiallergic and antibrowning molecules, as prooxidants, and even carcinogens. The type of food processing appears to determine which properties are produced. It has been observed that acrylamides are formed in many foods via the Maillard reaction at high temperature.
Reference
http://sphx.col.ynu.edu.cn/myfoodweb4/foodchemistry/maillard.html
Ames, J.M.; Dietary Maillard reaction products: Implications for human health and disease. Czech J. Food Sci. 2009, (27) S66-S69. http://www.agriculturejournals.cz/publicFiles/07583.pdf
Hodge, J. E. (1953). "Dehydrated Foods, Chemistry of Browning Reactions in Model Systems". Journal of Agricultural and Food Chemistry. 1 (15): 928–43. doi:10.1021/jf60015a004.
Hodge, J.E., Mills, F.D., and Fisher, B.E.; Compounds of browned flavor derived from sugar-amine reactions. Cereal Science Today, 1972, vol. 17, No. 2, 34-40. https://naldc.nal.usda.gov/download/31078/PDF.
Loudon, Marc: Organic Chemistry. 5th edition. Roberts and Company Publishers. 2009. http://www.macmillanlearning.com/Catalog/product/organicchemistry-sixthedition-loudon
Maillard LC. Action of amino acids on sugars. Formation of melanoidins in a methodical way. Compt Rend 1912; 154:66–68.
Tamanna, N., and Mahmood, N.; Food processing and Maillard reaction products: Effect on human health and nutrition. International Journal of Food Science. 2015. 1-6. https://www.hindawi.com/journals/ijfs/2015/526762/.
Zhang, Q., Ames, J.M., Smith, R. D., Baynes, J.W., and Metz, T. O.; A perspective on the Maillard reaction and the analysis of protein glycation by mass spectrometry: probing the pathogesis of chronic disease. J Proteome Res. 2009, 8(2): 754-769. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2642649/



.jpg)


.jpg)








