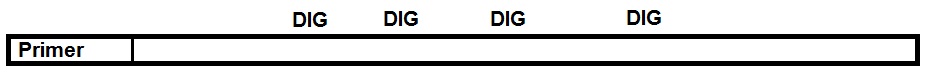Can single messenger RNAs (mRNA) be tracked inside live cells?
The answer is yes!
In recent years, single-cell biology has revealed that each cell is unique. However, single cells can vary significantly in their gene expression. Life is a dynamic process, and metabolic processes in cells are tightly regulated. The dynamics of RNA molecules including mRNAs can now be studied by tracking single RNA molecules. Recent advancements in fluorescence microscopy as well as in the synthesis of molecular probes now enable the study of cellular RNA dynamics. In 2006, Moon et al. reviewed the current state-of-the-art technology for tagging, delivery, and imaging useful for the tracking of single mRNA molecules in live cells.
Why is imaging of RNAs in living cells useful?
Imaging RNAs in cells allow studying the following events inside a living cell:
Transcription of RNA | Transcription of RNA is the initial step in gene expression and a regulation point for timing and production of gene products. |
Imaging of RNA Lifetime | Investigation of each gene expression step, starting from transcription to translation. Imaging RNA allows the study of gene expression from transcription to translation. However, imaging of proteins only provides information of the location of the end product of the expressed gene. |
Non-coding RNAs | Imaging and tracking RNA molecules also allow studying RNA molecules not translated into proteins. For example, the function of non-coding RNAs can be studied using labeled RNA. |
RNA counting | Single molecule analysis of RNA may enable counting numbers of mRNA molecules inside single cells. This should allow measuring gene expression levels in a quantitative manner. |
Ribonucleoprotein complexes | Single RNA imaging enables detection of subpopulations or transient states of messenger ribonucleoprotein (mRNP) complexes or particles. |
mRNPs | Tracking single mRNP molecules in real time may allow studying the sequence of events or RNA processing and transport. |
Having the ability to observe the organization and dynamics of RNA at the single molecule resolution in living cells will surely transform life science research in the near future.
How many molecules are there in a cell?
According to Moon et al. (2016), in chicken embryonic fibroblast cells there are approximately 2500 mRNA molecules present per cell, whereas there are approximately 108β-actin molecules present per cell. Therefore it is easier to count the number of RNA molecules in a single cell to study gene expression levels quantitatively than protein molecule numbers.
Labeling of mRNA
For tracking single particles organic dyes and fluorescent proteins are usually used for labeling mRNAs. However, hybridization probes or RNA motifs that bind to fluorescent molecules can also be used.
Oligodeoxynucleotide (ODN) probes
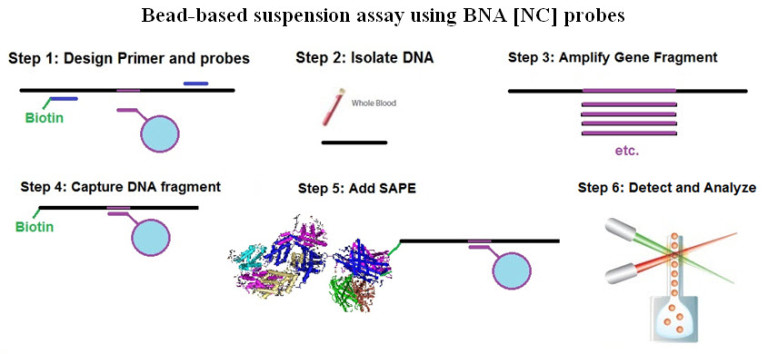
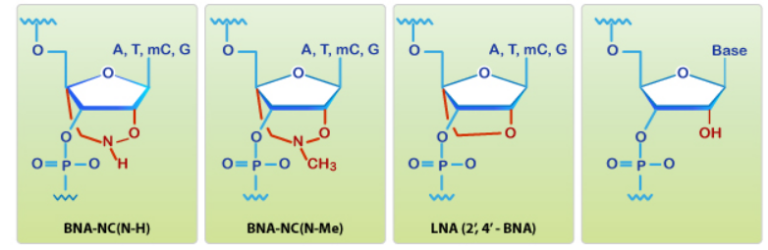
Target RNAs can be labeled via hybridization using short single-stranded DNA probes consisting of approximately 10 to 50 nucleotides. ODN probes are designed to be complementary to a target RNA sequence and are usually labeled with one or more fluorophore. To minimize background noise originating from free non-bound ODN probes, various strategies have been developed to only switch on fluorescence of the probes when bound to the target. This improves the signal to background ratio. To increase binding affinities to target sequences oligonucleotide mimics containing modified nucleotides are designed. Bridged Nucleotides (BNAs) can be used to enhance the stability and affinity of the probes.
Two main approaches are used for switching on the fluorescence signal: Förster resonance energy transfer (FRET) and static quenching.
Förster resonance energy transfer (FRET)
FRET refers to the radiationless transmission of energy from a donor molecule to an acceptor molecule. FRET occurs when two fluorophores are in proximity, approximately between 2 to 10 nm, and when the emission spectrum of the donor overlaps with the excitation spectrum of the acceptor. FRET can be used for sensitive detection of molecular interactions.
For FRET to work, two ODN probes are designed to hybridize to target RNA side-by-side. During hybridization, the donor and acceptor pair is brought together in the presence of the target (figure 1).
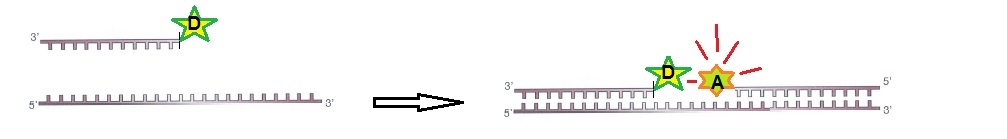
Figure 1: RNA ODN probes for FRET. Two designed ODN probes are hybridized to target RNA side-by-side. The fluorescence signal is switched on.
Static Quenching
Static quenching is a process in which fluorescence is decreased when the distance between the fluorophore and the quencher is less than 2 nm.
For static quenching ODN probes, one probe is labeled with a fluorophore and a second complementary ODN is labeled with a quencher molecule. The two labeled ODN probes are annealed together. In this configuration, the fluorophore and the quencher are close to each other resulting in no fluorescence. If the target RNA is present, the ODN is hybridized to the target with a higher affinity than to the second oligo. This reaction restores the fluorescence of the ODN labeled with the fluorophore. The observed fluorescence signal indicates that the target RNA is present. Hence, the presence of the target RNA results in a fluorescent signal originating from the ODN probe labeled with the fluorophore (figure 2).

Figure 2: Static quenching– Annealed ODN probes labeled with fluorophore and quencher.
Other probe types
Other probe designs are also possible. For example, fluorescently labeled oligonucleotide mimic probes can be designed using BNAs to enhance their hybridization affinity and stability.
A brief list is shown below:
Exiton-controlled hybridization-sensitive fluorescent oligonucleotide (ECHO) probes
Forced Intercalation (FIT) probes
Peptide nucleic acid and nano-graphene oxide (PANGO) probes
Sticky flare probes.
BNA probes contain nucleotide analogs that have a bridged structure in the sugar moiety. Optimal designed BNA probes increase base-discrimination, the stability of duplex or triplex formation, and show minimal cytotoxicity. These multi-functional synthetic RNA analogs can be spiked with DNA or RNA to modify structural formation of oligonucleotides. Because of their increased affinity to targets BNA based oligonucleotides enable detection of small or highly similar DNA or RNA targets.
Exiton-controlled hybridization-sensitive fluorescent oligonucleotide (ECHO) probes are designed to have thiazole orange (TO) dyes on a modified thymidine base. An exiton is defined as a bound state of an electron and an electron hole (electron-hole pair) that are attracted to each other via electrostatic Coulomb’s interaction. In other words, an exiton is an excited state formed by the recombination of an electron and an electron hole. During relaxation, the exiton gives off light and heat. Exitons are the main mechanism of light emission in semiconductors. Energy transfer processes occurring in exitons are radiative transfer, Förster transfer, and Dexter transfer. [https://en.wikipedia.org/wiki/Exciton].
FIT probes. Another probe type is a fluorescent probe designed as an oligonucleotide mimics containing dyes that replace one oligonucleotide in the middle of the probe. This type of probe is called a “forced intercalation probe” (FIT-probe). Oligonucleotide mimics labeled with asymmetric cyanine dyes, such as thiazole orange (TO), are forced intercalation probes that emit low background noise in the single-stranded state. When the probe is hybridized to the RNA target, the result is the intercalation of the TO dye within the duplex. The result is a strong fluorescence signal. A probe designed using peptide nucleic acid with a TO dye in the middle of the probe is an example for this. However, bridged nucleic acids (BNAs) can also be used for the design of these probes.
Sticky flare probes are made off 13nm gold particles functionalized with densely packed oligonucleotides. The gold core quenches the fluorescence of the target RNA. When the target RNA is recognized, sticky-flare transfers the fluorophore-conjugated ODNs to the RNA (figure 3). Sticky-flare probes can enter live cells by endocytosis. No transfection is needed. See also https://blog-biosyn.com/2013/08/29/what-are-nano-flares/.
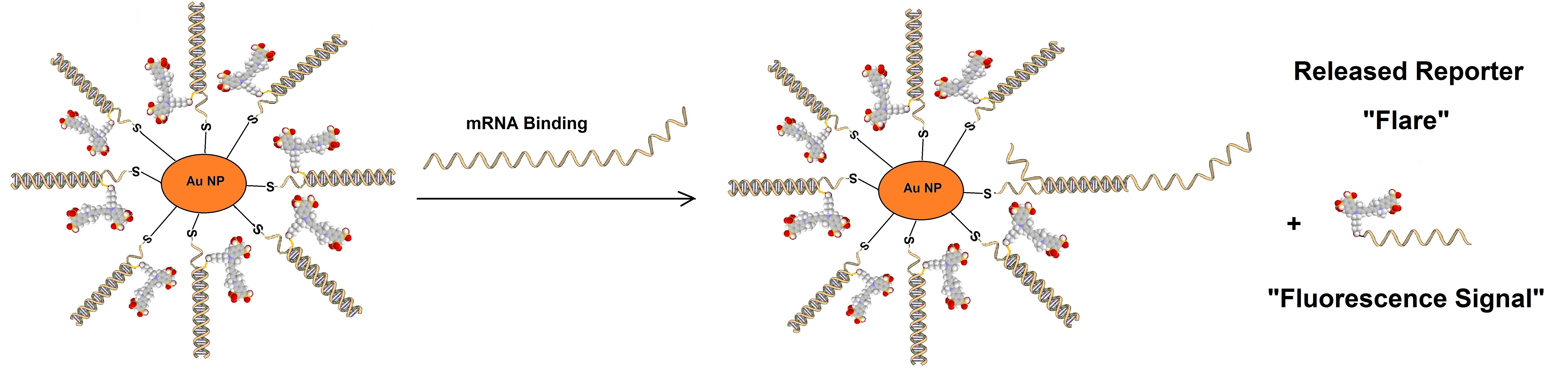
Figure 3: Sticky-flare or nano-flare based detection of mRNA.
Molecular Beacons
Molecular beacons are a type of ODN probes designed with a hairpin structure that forms a loop and a stem via self-complementary 5’ and 3’ arms. A fluorophore is attached to the end of one arm, and a quencher is attached to the end of the other arm (figure 4). Base pairing of the two arms keeps the fluorophore and the quencher in proximity which quenches the fluorescence. When the beacon encounters the target molecule containing a sequence complementary to the loop structure a probe-target hybrid is formed. This hybrid is energetically more stable than the self-complementary hairpin structure. After hybridization, the conformation of the beacon is changed, and fluorescence is restored. Since the beacon has no background signal in the absence of target molecules, higher signal-to-noise ratios are achieved in comparison to other ODNs.
Because of the advantages molecular beacons provide, they have already been used for the tracking and imaging of mRNAs of β-actin in fibroblasts, oskar in fruit fly oocytes, influenza virus mRNA in canine kidney epithelial cells, bovine respiratory syncytial virus RNA in bovine turbinate cells, and respiratory syncytial virus RNA in Vero cells.
![]()
Figure 4: Molecular Beacon based assay.
Furthermore, molecular beacons have a high level of specificity for target RNAs than linear ODNs, and they can be designed to allow to distinguish single nucleotide polymorphisms (SNP) in live cells. However, the melting temperature of the matching hybrids need to be above 37 °C, and the melting temperature of single nucleotide mismatch hybrids need to be below 37 °C.
Please review:
http://www.biosyn.com/moelcular-beacons.aspx,
http://www.biosyn.com/tew/molecular-beacon.aspx,
http://www.biosyn.com/tew/Design-rules-for-Molecular-Beacons.aspx
Tentacle Molecular Beacons
To achieve an even higher specificity tentacle molecular beacons with increased kinetics and affinity have also been developed. [https://www.ncbi.nlm.nih.gov/pmc/articles/PMC1904288/]. Tentacle probes are similar to molecular beacons but the presence of a capture region allows for enhanced specificity. Tentacle molecular beacons contain a hairpin structure similar to molecular beacons but are modified by the addition of a capture probe. Tentacle molecular beacons have increased kinetics and affinities. Usually, kinetic rate constants are up to 200-fold faster than that for molecular beacons with corresponding stem strengths.
Multi-color Molecular Beacons and Wavelength-Shifting Molecular Beacons
To allow simultaneous detection of multiple RNAs, multi-color molecular beacons and wavelength-shifting molecular beacons have also been developed. In the absence of targets, the probes do not fluoresce, however, when the targets are encountered the probes usually fluoresce in the emission range of the emitter fluorophore. Wavelength-shifting molecular beacons are brighter than conventional molecular beacons. [http://www.nature.com/nbt/journal/v18/n11/full/nbt1100_1191.html]
Dual FRET Molecular Beacons
To overcome false-positive signals of conventional molecular beacons, dual FRET molecular beacons have been designed. Dual FRET probes can achieve higher signal-to-noise ratios than can single molecular beacons.
Light Induced Molecular Beacons
Light-induced molecular beacons can hybridize to their target only when activated with UV light. This approach permits the fine control of timing and location of RNA labeling. Molecular beacons can be synthesized with caged nucleobases in the loop region. The resulting constructs remain non-fluorescent in the presence of the target RNA. The exposure to light (366 or 405 nm) in vitro or in cells fully activates the beacons. Molecular beacons synthesized with the caged nucleobases dANPE, dCNPE, or sGNPP cannot form normal Watson-Crick base pairs. However, after irradiation with light, the photo-labile caging groups are removed, and the unmodified nucleobases are regenerated. This restores the ability for base pairing of the molecular beacon resulting in a fluorescent signal of the probe-target hybrid.
[http://www.biosyn.com/tew/Light-sensitive-nucleotides.aspx, http://pubs.rsc.org/is/content/articlehtml/2012/cc/c2cc16654b,
Avoiding sequestering of molecular beacons into the nucleus
Sometimes molecular beacons tend to sequester into the nucleus, which can cause a nonspecific fluorescent signal. To prevent this, large proteins or nanoparticles have been attached to molecular beacons to prevent the passing of the beacon through the nuclear pores.
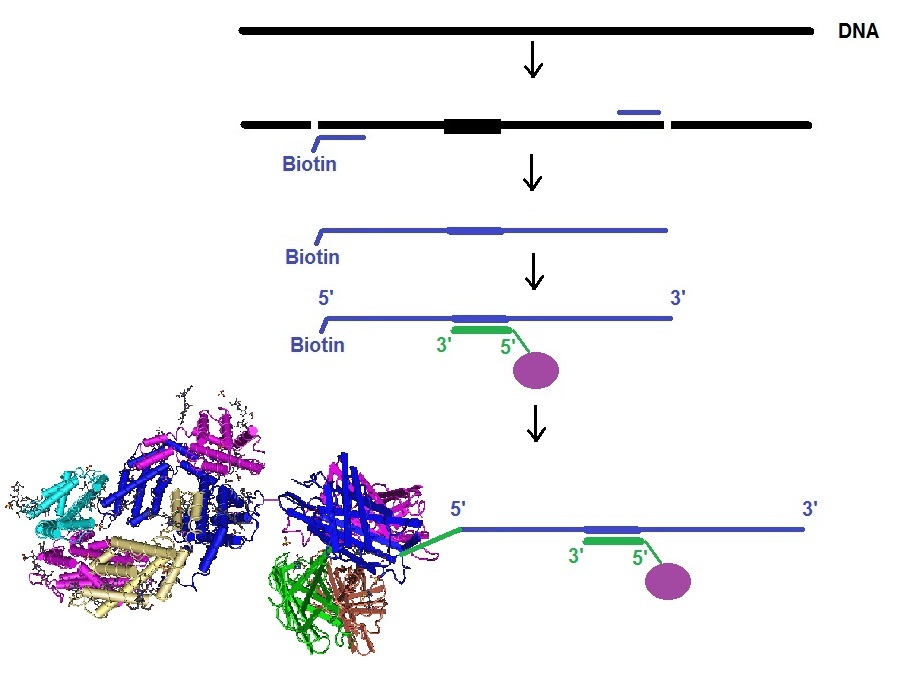
RNA Stem-Loop system
In this approach, mRNA is labeled in living cells using RNA stem-loop motifs. The MS2 bacteriophage coat protein (MCP) is known to exhibit strong affinity for the unique RNA stem-loop sequence MS2 binding site (MBS).[https://www.ncbi.nlm.nih.gov/pmc/articles/PMC413242/]. The MBS stem-loop sequence is a short oligonucleotide sequence, containing approximately 20 nucleotides. Because of this, multiple MBS stem-loops can be used for the tagging of the mRNA of interest. The signal of a single mRNA can be amplified by increasing the number of MBS stem-loops. Each MBS stem-loop can bind a dimer of MCP fused to a fluorescent protein (FB). With this approach, a single copy of mRNA can be labeled with many FBs. Other similar stem-loops such as the PP7 bacteriophage system and the λ-phage N system can also be used.
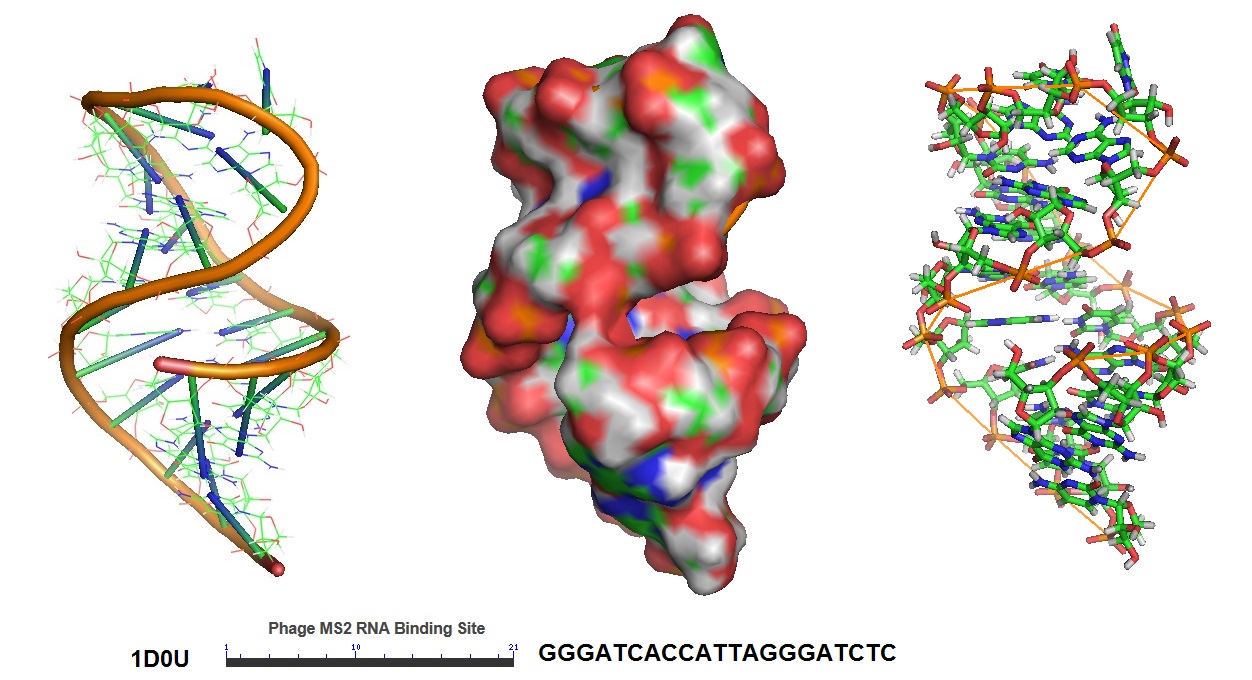
Figure 5: Structural model of the phage MS2 RNA hairpin-loop binding site.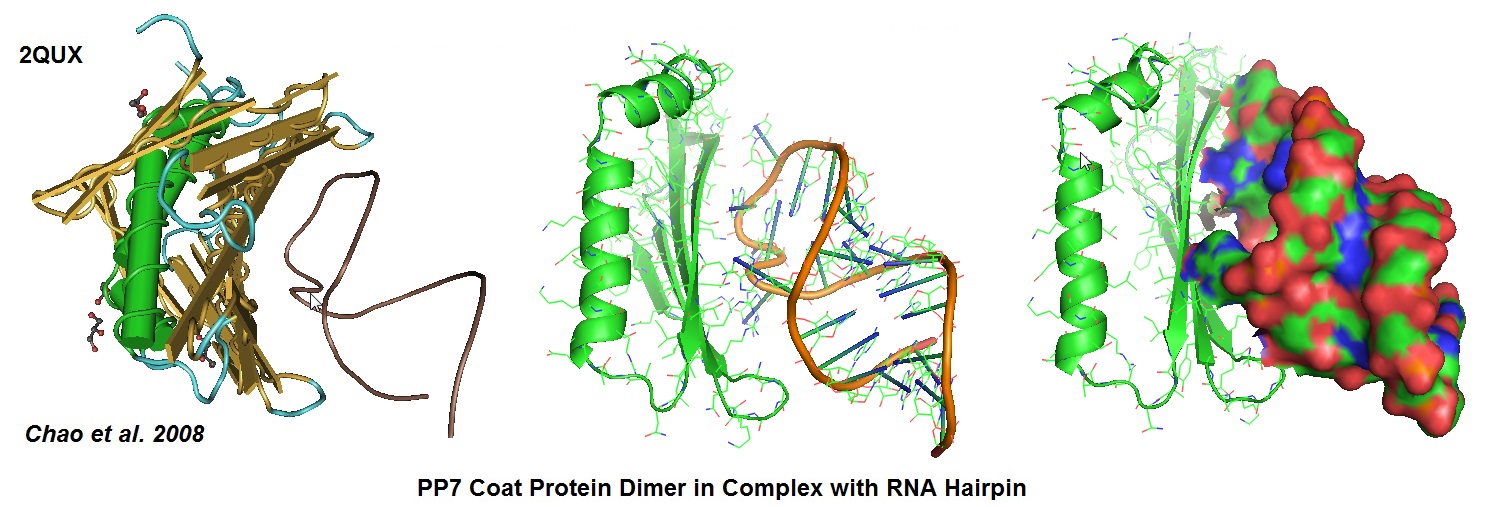
Figure 6: Structural models derived from the cocrystal structure of the PP7 bacteriophage coat protein in complex with its translational operator (Chao et al. 2008). The structure illustrated the molecular basis of the PP7 coat protein’s selective binding to the cognate RNA. The conserved beta-sheet surface recognizes the RNA hairpin. Different depictions are used for the image.
Aptamer-fluorogenic System
An aptamer called “Spinach” was developed to bind and activate the fluorogen, 3,5-difluoro-4-hydroxy-benzylidend imidazolinone (DFHBI). DFHBI is a derivative of the green fluorescence protein’s (GFP) fluorphore 4-hydroxybenzlidine imidazolinone (HBI). This RNA aptamer induces fluorescence of a GFP-like chromophore.
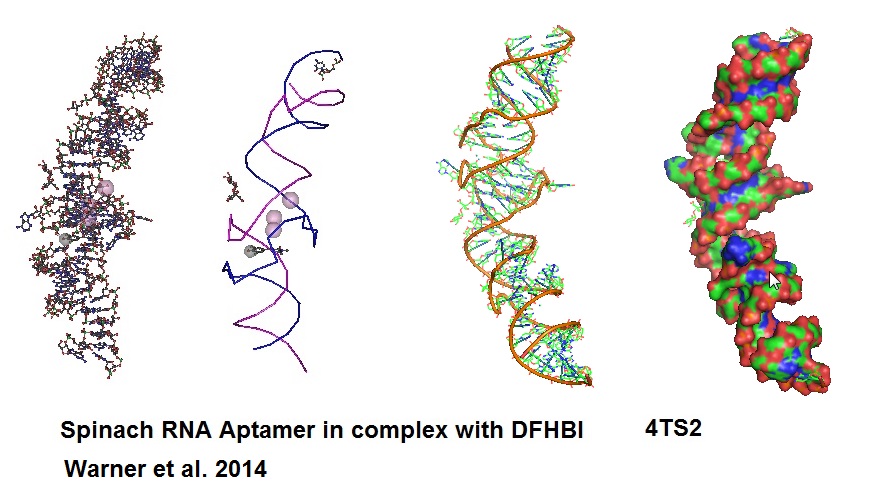
Figure 7: Structural models of RNA Aptamer Spinach.
When Spinach binds to DFHBI a Spinach-DFHBI complex is formed which emits fluorescence. Molecular modules based on the Spinach sequence can be designed for the detection of other cellular molecules. New aptamers are constantly developed to enable investigation of a variety of molecules found in cells. However, because of some thermal instabilities and misfolding tendencies of aptamers when expressed or injected into living cells, aptamers with enhanced folding properties will need to be designed. Advance protocols using Systematic Evolution of Ligands by Exponential Enrichment (SELEX) can be coupled with fluorescence-activated cell sorting (FACS) for the development of brighter RNA aptamer-fluorogenic systems.
Glossary
Å Ångström: 1 Å = 0.1 nm
Broccoli Newer aptamer
DFHBI 3,5-difluoro-4-hydroxy-benzylidene
ECHO Exciton-controlled hybridization-sensitive oligonucleotide
FIT Forced intercalation
FRET Förster resonance energy transfer
GFP Green Fluorescence Protein
HBI 4-hydroxybenzlidene imidazolinone
MBS MS2 binding protein
MCP MS2 bacteriophage coat protein
mRNA Messenger RNA
mRNP Messenger riponucleoprotein
ODN Oligodeoxynucleotide
PANGO Peptide nucleic acid nano-graphene oxide
TO Thiazole ornage
SNR Signal-to-noise ratio
Spinach RNA aptamer specifically binding to DFHBI
Sticky-flare Functionalized gold particle with densely packed oligonucleotides
Appendix
Table 1: Average Bond Lenghts
Bond Bond length (Å) Bond Bond length (Å)
C-C 1.54 N-N 1.47
C=C 1.34 N=N 1.24
C≡C 1.20 N≡N 1.10
C-N 1.43 N-O 1.36
C=N 1.38 N=O 1.22
C≡N 1.16
C-O 1.43 O-O 1.48
C=O 1.23 O=O 1.21
C≡O 1.13
Reference
Chao JA, Patskovsky Y, Almo SC, Singer RH; Structural basis for the coevolution of a viral RNA-protein complex. Nat.Struct.Mol.Biol. (2008) 15 p.103.
Hyungseok C Moon, Byung Hun Lee, Kiseong Lim, Jae Seok Son, Minho S Song and Hye Yoon Park; TOPICAL REVIEW- Tracking single mRNA molecules in live cells. Journal of Physics D: Applied Physics, Volume 49, (2016) Number 23.
Khashti Ballabh Joshi, Andreas Vlachos, Vera Mikat, Thomas Deller and Alexander Heckel; Light-activatable molecular beacons with a caged loop sequence. DOI: 10.1039/C2CC16654B (Communication) Chem. Commun., 2012, 48, 2746-2748.
Santangelo PJ, Nix B, Tsourkas A, Bao G.; Dual FRET molecular beacons for mRNA detection in living cells. Nucleic Acids Res. 2004 Apr 14;32(6):e57.
Brent C. Satterfield, Jay A.A. West, and Michael R. Caplan; Tentacle probes: eliminating false positives without sacrificing sensitivity. Nucleic Acids Res. 2007 May; 35(10): e76. PMCID: PMC1904288.
Smith JS, Nikonowicz EP.; Phosphorothioate substitution can substantially alter RNA conformation. Biochemistry. 2000 May 16;39(19):5642-52. RNA hairpin containing the binding sitwe for bacteriophage MS2 capsid protein.
Katherine Deigan Warner, Michael C. Chen, Wenjiao Song, Rita L. Strack, Andrea Thorn, Samie R. Jaffrey, and Adrian R. Ferré-D’Amaré; Structural basis for activity of highly efficient RNA mimics of green fluorescent protein. Nat Struct Mol Biol. 2014 Aug; 21(8): 658–663. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4143336/.
--...--
.png)