The Chemical Synthesis of OligonucleotidesBy Andrei Laikhter and Klaus D. LinseThe study of nucleic acids has now become a fruitful and dynamic scientific enterprise. Nucleic acids are of unique importance in biological systems because genes are the fundamental unit of heredity. The process how genes are expressed in all living organisms is fundamentally important and most genes are located in the chromosomes within the cell nucleus. Genes express themselves via a protein machinery in the cytoplasm. The genetic material was identified as deoxyribonucleic acid (DNA) in 1944 (Avery et al. 1944). Next, the double-helical nature of DNA was revealed in 1953 by Francis Crick, James Watson and Maurice Wilkins. Furthermore, the combined action of multiple genes defines the properties or phenotype of higher organisms. Even to this date multi-gene characteristics are difficult to analyze. Genes are made up of deoxyribonucleic acid or DNA, and each gene is a linear segment, or polymer, of a long DNA molecule. However, genes have been found to be nonrandomly distributed on the chromosomes and vary enormously in size and intron-exon structure. A DNA polymer, or DNA oligonucleotide, contains a linear arrangement of subunits called nucleotides. There are four types of nucleotides. Each nucleotide has three components; a phosphate group, a sugar and a base that contains nitrogen within its structure. The sugar moiety in DNA oligonucleotides is always dexoyribose, and there are four alternative bases: adenine (Ade, A), thymine (Thy, T), guanine (Gua, G), and cytosine (Cyt, C). The phosphate groups and the deoxyribose sugars form the backbone of each DNA stand. The bases are joined to the deoxyribose sugar and stick out to the side. Both, DNA and ribonucleic acid (RNA), consist of 5’-3’ phosphodiester-linked nucleotide units that are composed of a 2’-deoxy-D-ribose (DNA) or D-ribose (RNA) in their furanose forms and a heteroaromatic nucleobase (A, T, G, and C; A, U, G, C), and the resulting oligonucleotide chain is composed of a polar, negatively charged sugar-phosphate backbone and an array of hydrophobic nucleobases. The amphiphilic nature of these polymers dictates the assembly and maintenance of secondary and tertiary structures the oligonucleotides can form. In double stranded DNA, the bases of one strand are paired with the bases in the other strand. Adenine (A) in one strand is paired with thymine (T) in the other strand and guanine (G) in one strand is paired with cytosine (C) in the other strand as well. In the DNA duplex structure, genetic information is stored as a linear nucleotide code. This code can be accessed and replicated. RNA, or ribonucleic acid, is another structurally related essential biopolymer. RNA differs from DNA in having the sugar ribose in place of the deoxyribose. Furthermore, in RNA the thymine (T) is replaced with uracil (U). The bases A and G are purine bases as they contain a double ring structure called the purine ring. The other two bases, C and T, are pyrimidine bases because they contain a single pyrimidine ring. Hydrogen bonds hold the two strands together. 

Figure 1: Structures of nucleic acids. Figure 2: Atomic numbering and definitions of torsion angles for one nucleotide according to the UPAC nomenclature. The designation of chain direction and main chain atoms of i th unit in a polynucleotide chain and the atom numbering for the bases of common nucleosides and nucleotides are illustrated. Hydrogen atoms carry the same numbers as the heavy atoms to which they are attached. The name in parenthesis applies when the 'd' in parenthesis in the formula is present. (Source: www.chem.qmul.ac.uk/iupac/misc/pnuc1.html). 
Figure 3: Representative types of base pairs. Purines and pyrimidines can form base pairs through hydrogen bonds. Watson-Crick, Hoogsten and wobble base pairs are the most common. An extensive list of graphical base pairs can be found in “Wolfram Sanger: Principles of Nucleic-Acid Structure. Spinger-Verlag New York Berlin Heidelberg Tokyo, pp 120.” In biological systems, nucleic acids exist in higher ordered structures held together through self-assembly. Since hydrogen bonding and base stacking are the major driving force for this self-assembly the structural properties of nucleotides can affect the self-assembly process. These molecular forces dictate the final tertiary structure of nucleic acids (The RNA World). The central dogma of molecular biology, originally described by Francis Crick, illustrates how DNA is transcribed into RNA which is ultimately translated into protein sequences. According to the dogma nucleic acid alone may store information or specify the sequence of gene products, proteins. Proteins are never able to specify a nucleic acid or protein sequence.  Figure 4: The central dogma of molecular biology.  Figure 5: Structures of oligonucleotides. In the beginning years, the chemical synthesis of specific oligoribo- and oligodeoxyribonuclotides has been regarded as a somewhat esoteric chemistry of natural compounds. The advent of gene technology and the development of new chemical and analytical methods such as high performance liquid chromatography (HPLC), 31P-NMR spectroscopy and automated synthesis methods has changed this all. The development and introduction of commercially available “gene synthesizing machines” following the development of chemical DNA and RNA synthesis strategies has made the synthesis of natural and artificial oligo-nucleotides a routine procedure as well as more cost-efficient and faster. To allow for a successful oligonucleotide synthesis the following fundamental prerequisites need to be established: - All reagents should be soluble in non-aqueous solvents.
- The amino and hydroxyl functions of nucleotide bases and sugar residues must be suitably blocked.
- Protecting groups, introduced during synthesis, must be stable under conditions of chain elongation when the internucleotide phosphodiester bonds are formed.
- Protecting groups should be labile enough to allow removal at the end of the synthesis without damaging the reaction products.
- Final yields of the synthesis should be almost quantitative to avoid products with failure sequences.
Several strategies have been developed in the past which comply with these limiting provisions. The phosphodiester and the phosphotriester approaches, both, utilize protection of the 3’ and 5’ hydroxyl groups of the deoxyribose. The phosphotriester uses a third protecting group for the protection of the internucleotide bond. A third approach called the “phosphate” procedure employs compounds with trivalent phosphorus and can be regarded as a trimester method. The development of oligonucleotide synthesis started with the phosophotriester method in 1955 but the fist significant success is reported to be achieved using the phosphodiester method when the genes for alanine and tyrosine suppressor tRNAs of yeast and E. coli were synthesized in the 1980s. Figure 6: Outline of the synthesis of the dinucleotide d(TpT) by Michelson and Todd reported in 1955. In this approach, 3’-O-acetylthymidine was phosphorylated with a phosphorochloridate, and the phosphate group was protected with a benzyl group. The structure of the dinucleotide was confirmed by enzymatic digestion (Michelson & Todd, 1955). This synthesis approach later became known as the phosphotriester approach. The literature reports that Gobind Khorana, in 1956, accidentally discovered the phosphodiester method for the chemical synthesis of deoxyribo-oligonucleotides (Khorana et al., 1956). The exploitation and further development of this method by many scientists in subsequent years for the chemical synthesis of deoxyribo-oligonucleotides led to the elucitation of the genetic code and the first total synthesis of a gene (Khorana et al., 1956; Khorana, 1961, 1969, 1979). Marshall Nirenberg and Gobind Khorana broke the genetic code and could assign code words called codons. Codons are triplets of nucleotides coding for the twenty amino acids. Both scientists received the Nobel Prize in Physiology or Medicine in 1968 together with Robert Holley. Phosphotriester Method Where B = A,C, G or T nucleo bases; R1, R2, R3 = protecting groups; X = Halogen Figure 7: General outline of the phosphotriester method. Figure 8: Outline of Khorana’s synthesis approach later known as the phosphodiester approach. In this approach, 5’-O-tritylthymidine and 3’-O-acetylthymidine 5’-phosphate are reacted in the presence of toluene-4-sulfonyl chloride (TsCl) or N1,N3-dicyclohexylcarbodiimide (DCC) in a pyridine solution. The removal of the trityl and acetyl group yields the d(TpT) dinucleotide (Khorana et al., 1956; 1957; Gilman and Khorana, 1958). Posphodiester Method Where B = A,C, G or T nucleo bases; R1, R2 = protecting groups Figure 9: General outline of the phosphodiester method.In the phosphodiester method the phosphate group between the two nucleotides is unprotected which makes the resulting compounds only soluble in organic solvents to a limited extent. Phosphite Method Where B = A,C, G or T nucleo bases; R1, R2, R3 = protecting groups; X = Halogen, N(CH3)2, morpholine Figure 10: Chemistry of phosphite method.Willi Bannwarth in 1985 reported a simple synthesis of phosphoramidite dinucleotides with two different phosphorous-protecting groups for the synthesis of 2′-oligodeoxynucleotides on a polymer support called the “Phosphite-Triester Method. Even though oligonucleotides can be assembled manually in a step-wise fashion this process has been and is quite laborious and demanding. The availability of commercial DNA synthesizers has made the process easier and more cost effective. The principle of solid phase synthesis was first developed and applied to the synthesis of polypeptides by Robert Bruce Merrifield (July 15, 1921 – May 14, 2006), an American biochemist who won the Nobel Prize in Chemistry in 1984 for the invention of solid phase peptide synthesis. He realized that the key to a successful synthesis is to anchor the first monomer to an insoluble polymeric support. Other monomers can then be joined, one by one, to the fixed terminal end of the growing polymer. At the end of the synthesis, the completed polymer chain can be detached from the insoluble polymer and purified. This process has been further optimized over the years to become highly efficient and has now become a fundamentally important method employed in automated oligonucleotide synthesizers. I. General Methods of Solid Phase Oligonucleotide Synthesis.a) Phosphoramidite method The phosphoramidite method of DNA synthesis is currently considered as the standard synthesis method used in most automated synthesizers today. This method allows achieving the high coupling efficiencies needed to synthesize longer and longer oligonucleotides with low amounts of failure sequences. The oligonucleotide phosphoramidite synthesis chemistry was introduced nearly 20 years ago (McBride and Caruthers, 1983). Building blocks used for synthesis are commonly referred to as “monomers”, which are activated DNA nucleosides (phosphoramidites). The dimethoxytrityl (DMT) group is used to protect the 5’-end of the nucleoside, a β-cyanoethyl group protects the 3’-phosphite moiety, and may also include additional groups that serve to protect reactive primary amines in the heterocyclic nucleo bases. The protecting groups are selected to prevent branching or other undesirable side reactions from occurring during synthesis. Oligonucleotides are synthesized on solid supports. Typically, the support is a small column filled with control pore glass (CPG), polystyrene or a membrane. The oligonucleotide is usually synthesized from the 3’ to the 5’. The synthesis begins with the addition of a reaction column loaded with the initial support-bound protected nucleotide into the column holder of the synthesizer. The first nucleotide building block or monomer is usually anchored to a long chain alkylamine-controlled pore glass (LCAA-CPG). A schematic diagram general outline the solid phase oligonucleotide synthesis of a dinucleotide is illustrated below.  Figure 11: Diagram of the workflow for the phosphoramidite method. Figure 11: Diagram of the workflow for the phosphoramidite method.The phosphoramidite approach to oligonucleotide synthesis proceeds in four (4) steps, which are schematically outlined in figure 11. Automated synthesis is done on solid support, usually controlled pore glass (CPG) or polystyrene. Synthesis is initiated with cleavage of the 5’-trityl group by brief treatment with dichloroacetic acid (DCA) dissolved in dichloromethane (DCM). Next, the monomer activated with tetrazole is coupled to the available 5’-hydroxyl resulting in a phosphite linkage. Subsequent phosphite oxidation by treatment with iodine using a THF/pyridine /H2O solution yields a phosphate backbone. The capping step with acetic anhydride, which terminates undesired failure sequences, completes the cycle of oligonucleotide synthesis. As figure 12 illustrates a typical synthesis cycle includes a condensation, a capping, an oxidation, and a cleavage or deprotection step. In general, automated DNA oligonucleotide synthesis produces a single-stranded oligonucleotide product per column.  Figure 12: A summary of the Phosphoamidite Oligonucleotide Solid Phase Synthesis Method. Removal from the support and final base deprotection process Figure 12: A summary of the Phosphoamidite Oligonucleotide Solid Phase Synthesis Method. Removal from the support and final base deprotection processAfter the final sequence has been assembled, the oligomer must be removed by cleaving it from the support and fully deprotected prior to its use. A 90 minute treatment with ammonium hydroxide at room temperature can be used to cleave the oligomer from the support and to deprotect the phosphorous by β–elimination of the cyanoethyl group. The acetyl capping groups and the base protecting groups are more difficult to remove and a 24 hour treatment at room temperature or an overnight treatment at 55 °C with ammonium hydroxide allows for effective removal of these groups. After cleavage and deprotection, the resulting crude mixture contains the product oligomer, possible truncated failure sequences with free 5’hydroxyl ends, byproducts of deprotection, and silicates from hydrolysis of the glass support. Different purification methods can be used to separate the product oligonucleotide from the contaminating species. b) H-Phosphonate method. The first internucleotide reactions employing H-phosphonates were reported in the 1950s (Hall et al., 1957), and the first solid supported oligonucleotide synthesis was described in the early 1970s (Kabachnik et al., 1971), where the reported coupling efficiency was between 46 and 80%. The modern H-phosphonate method involves the use of triethylamine (TEA) or 1,8-Diazabicyclo[5.4.0]undec-7-ene salts, or DBU salts of corresponding 3’-H-phosphonate monomers, which need to be activated with the appropriate acetyl chloride followed by coupling of activated O-acetyl phosphonate to the 5’-hydroxyl of the previous nucleoside attached to solid support (Figure 13). Isopropyl-H-phosphonate together with the same activator is used for the capping step of the cycle.  Figure 13: Diagram depicting the H-Phophonate method. Figure 13: Diagram depicting the H-Phophonate method. The resulting poly-H-phosphonate backbone can be oxidized after the final oligonucleotide is assembled from solid support in one step. The coupling efficiency of H-phosphonate synthesis is usually 94-95%, which is quite low compared to the phosphoramidite method. This approach is mainly used in cases when the desired modification of the phosphorous backbone can be done only through an H-phosphonate intermediate such as by using boranophosphates (Sergueev & Shaw, 1998) or phosphoramidates (Froehler, 1986), or others. c) Phosphotriester method. The phosphotriester approach (Letsinger & Mahadevan, 1965, 1966) is another successful method of oligonucleotide synthesis on solid support. Usually the coupling phosphorylating agent is an activated HOBt phosphate ester (Figure 14).  Figure 14: Diagram of the phosphotriester approach. Figure 14: Diagram of the phosphotriester approach.II. RNA synthesis.Automated RNA synthesis on solid support was introduced a decade later than DNA synthesis and many disappointing results had to be overcome leading to a difficult history of development. The use of one extra 2’-hydroxy group makes this chemistry magnitudes more difficult compared to conventional DNA synthesis. The protective group of 2’ hydroxyl in ribose must remain intact both during the synthesis and base deprotection step. The last property of the protection is critical due to the hydrolytic instability of RNA in basic conditions. That is the reason why all modern methods of RNA synthesis that employ different RNA phosphoramidite monomers have protective groups at the 2’ hydroxyl group. This protective group needs to be stable in basic conditions during base deprotection and can be easily removed to completion on the next processing step using different or orthogonal conditions. TBDMS Phosphoramidites TBDMS Phosphoramidites are the first type of RNA monomers that have been developed by Kelvin Ogilvie group at McGill University in mid 1980s. That type of monomers has a 2’- tert-butyldimethylsilyl (TBDMS) protecting group and it is the most common type of building blocks used in solid phase RNA synthesis. The protecting group is stable in methylamine solution and less stable in ammonia. Disadvantage : The phosphoramidite building blocks require longer coupling times compared to other RNA building blocks. | 
Figure 15: TBDMS phophoramidites | TOM phosphoramidites Xeragon AG introduced TOM phosphoramidites about six years ago. This type of monomer has a 2’-triisopropyl-silyl-oximethyl (TOM) protecting group, which is quickly removed by treatment with tetrabutylammonium fluoride (TBAF). The biggest benefit of this chemistry is that it allows the synthesis of longer up to 100 mer oligonucleotides. Disadvantage :It is commercially available only for research applications. | 
Figure 16: TOM phophoramidites | Fpmp Phosphoramidites Fpmp Phosphoramidites have 2’-fluorophenyl-methoxy-pipiridyl (Fpmp) protection groups. These phophoramidites were developed by C. B. Reese in the late 1980s and the first commercially available RNA phosphoramidites were introduced by Cruachem Ltd. in the early 1990s. One of the main advantages of this chemistry is that the protecting group is useful for RP HPLC and stays intact until all post-synthetic manipulations are done. Most importantly this protection group prevents RNA from cleavage by RNases. Disadvantage :It is commercially available only for research applications. | 
Figure 17: Fpmp phophoramidites | ACE Monomers ACE monomers were developed in M. Caruthers’s group and introduced in the mid 1990s by Dharmacon, Inc. This type of RNA monomers have a 2’-acetylethoxy orthoester (ACE) protecting group and are 5’-hydroxyl protected with a silyl protecting group. This approach differs from conventional DMT protection. The ACE orthoester group is removable using mild acidic conditions. In order to avoid complications as observed in the case of Fpmp chemistry, ACE monomers have been designed with special 5’ silyl protecting groups that are removable in the presence of tetra-n-butylammonium fluoride (CH3CH2CH2CH2)4N+F-) or TBAF. Disadvantage :Contrary to DMT, 5’-silyl protection is not a convenient colored marker and cannot be used for quantitative and qualitative synthesis evaluation. | 
Figure 18: ACE monomers |
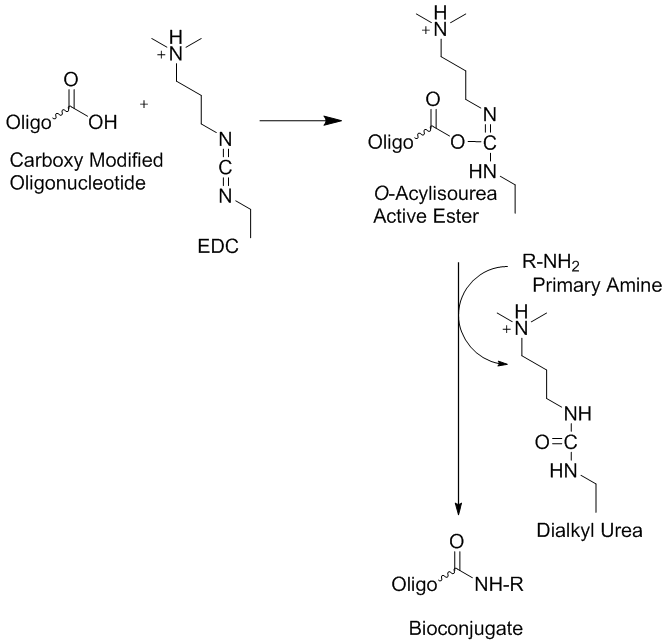
III. Synthesis of modified oligonucleotidesThe synthesis of modified oligonucleotides involves the selection of the best strategy to design and plan the needed synthetic approach or pathway. The following paragraph shows how to develop a synthetic strategy and introduce desired modification into oligonucleotide using manual phosphoramidite coupling and how to optimize post synthetic labeling with an activated ester. a) Strategy of synthesis A good way to plan a synthesis is to start with a retro-synthetic analysis. This approach starts with determining what the final product should be and going backwards along the synthetic route to determine what kind of reagents have the appropriate protecting groups needed for the synthesis. An example is shown below. The synthetic path needs to be designed using modifications that do not interfere with other functional groups. That is, the best orthogonal conditions will need to be established. b) On-Support modifications The easiest on support modifications known is the oxidation of H-phosphonates or phosphite triesters using elemental sulfur or disulfides to afford phosporothioates. There are a number of modifications of the phosphate backbone, which can be done while the oligonucleotide is attached to solid support. Most of them require special phosphoramidites, such as the use of alkyl phosphonates and phosphate triesters, or others. Some of them require an H-phosphonate backbone for the transformation. Examples are the use of boranophosphates (Sergueev & Shaw, 1998) or phosphoramidates (Froehler, 1986). Internal modifications on oligonucleotides can be done using either corresponding extendible amidites or using an asymmetrical branch with a non-extendible amidites and any 5’ modifications can be introduced using any type of monomers using an automated synthesizer. In addition any 3’ modification can be introduced using the appropriate solid support. The reversed synthesis from the 5’ to 3’ end can be employed for 3’ modifications if a solid support is not available but the needed phosphoramidite is. The same approach is applicable for 5’ modifications when the modifications are only available on a solid support. Furthermore, phosphoramidite chemistry is not the only chemistry that can be performed using solid support. Some peptide chemistry methods are also compatible with oligonucleotide synthesis methods. For example NHS esters or similar activated esters can be attached to unblocked aliphatic amino-linkers on solid support. In this case the desired molecular probe or reporter molecule has to be stable in basic conditions used during cleavage and base deprotection. c) Post-synthetic modifications. The most common chemo-selective reagents for post synthetic oligonucleotide modifications, for example to introduce different molecular probes and markers, are N-hydroxysuccininidyl activated esters. This type of activated esters reacts very selectively with aliphatic primary amines and is very stable in aqueous buffers at pH 8-9 towards hydrolysis. However, the following three very important issues in post-synthetic NHS labeling need to be addressed. First, the pH of the reaction mixture needs to be maintained at pH 8-9 during the coupling reaction. Second, the correct salt form of the oligonucleotide needs to be used, since ammonia or amino contaminants can also react with NHS esters to form unwanted different by-products. Therefore it is very important to exchange all ammonia ions with other types of counter ions such as lithium or sodium ions. This can be achieved either by precipitation of the oligonucleotides with lithium or sodium ions or by using HPLC with the corresponding buffers. Third, another important requirement for a successful reaction is to make sure that the NHS ester is completely soluble in the reaction mixture of the corresponding buffer and DMSO or other organic solvent suitable for the reaction used. This is needed because the precipitated ester is unreactive. This method can be used not only for conjugation of DNA to small molecules, but also for DNA-protein conjugations (Jablonski et al. 1986). Michael addition is another useful selective reaction for oligonucleotide post synthetic modifications. Maleimide derivatives and sulfhydryl modified oligonucleotides are usually employed for this method. This approach has been used for conjugation DNA and proteins as well (Ransom Hill Bioscience, Inc, Technical Bulletin). There are a few other chemoselective methods, which have been used for DNA–protein conjugations and the attachment of modified oligonucleotides to solid supports, such as those involving aldehydes and hydrazides, disulfides and thioesters, etc. (see IDT Technical Bulletins). All these methods are very useful for the introduction of post-synthetic oligonucleotide modifications. d) Automated DNA/RNA synthesizer One of the earliest, or the earliest, DNA/RNA synthesizer can be viewed at the Smithsonian Institution in Washington DC. The DNA/RNA synthesizer model 394 is made out of steel, glass, plastic and insulated wire. It has the measurements: 50 cm x 66 cm x 46 cm; 19 11/16 in x 26 in x 18 1/8 in. This Model from Applied Biosystems, Inc. was on the market from 1991 to 2007. DNA/RNA synthesizers can produce short single strands of nucleotides known as oligonucleotides which can be further linked together to create longer strands of DNA or RNA. Because of the automated approach it is now often easier to make stretches of DNA or RNA with a synthesizer than it is to isolate the same stretches of DNA or RNA from natural sources. In addition, synthetic oligonucleotides can also be created with slight changes from the naturally occurring forms, such as the insertion of artificial nucleotides, allowing researchers to study the impact of modified molecules. | ![]() Figure 21: DNA/RNA synthesizer model 394. Figure 21: DNA/RNA synthesizer model 394. |
Figure 22 shows pictures of typical reaction columns containing either activated or monomer loaded CPG supports as well as pictures of automated oligonucleotide synthesizers. Using this type of supports succinylated oligonucleotides or their monomers can be coupled to aminophenyl- or aminopropyl-derivatized glass surfaces, and disulfide-modified oligonuclotides or their monomers can be immobilized onto a mercaptosilanized glass support using a thiol/disulfide exchange reaction or through chemical cross-linkers. Figure 22: Icons of reaction columns and automated oligonucleotide synthesizers are shown. (Left) Different types of columns, color coded. (Middle) Columns mounted into the holder of the synthesizer. (Right) ABI 3900 and Expedite High-Throughput DNA/Oligo Synthesizers. Table 1: Some milestone in DNA/RNA solid phase oligonucleotide synthesis.| Year / decade | Event | Researcher(s) | | 1955 | First published account of the chemical synthesis of an oligonucleotide. Synthesis of a dithymidine dinucleotide containing a 3′: 5′-internucleotidic linkage. Phosphotriester method. | Michelson and Todd | | 1958 | Improved and general method for the synthesis of ribo-and deoxyribo-nucleoside-5' triphosphates. Khorana and co~workers first used the tetrahydropyranyl group (Thp) as 2' -protection in the synthesis of mononucleotides. | Smith and Khorana | 1962
1963 | On-off protection for sequential oligonucleotide synthsis.Stable phophorylated nucleosides are coupled to desired nucleoside when activated. | Korana lab
Smith et al.
Schaller et al. | | 1960s | Robert Letsinger devised a method for assembling oligos via solid phase chemistry. Oligonucleotides are constructed by linking chemical building blocks onto polymer beads. This, together with slight adjustments to Khorana’s original protocol, simplified the reaction so that the first automated machines to perform oligonucleotide synthesis could be built in the late 1970s. | Letsinger | | 1963 | Stepwise synthesis of ribooligonucleotides. | Lapidot & Khorana | | 1965, 1966 | Phosphotriester approach | Lapidot & Khorana | | 1980s | Further adjustments to the reaction made it possible for someone without a great deal of experience in chemical preparation to operate the machines, opening up their use to a wider audience and increasing their commercial viability. | | | 1985 | RNA synthesis using 2'-O-(tert-butyldimethylsilyl) protection | Ogilvie | | 1986 | H-Phosphonate Diester Intermediates | Froehler | | 1989 | Applied Biosystems started marketing oligonucleotide synthesizers. | ABI | | 1998 | H-Phosphonate Approach | Sergueev | | 1991 to 2007 | DNA/RNA synthesizer model 394. This model is considered as the second wave of Applied Biosystems’s DNA/RNA synthesizers which consumed chemicals more efficiently than the previous model and could synthesize up to four oligos at one time. | ABI |
Reference Abbreviations and Symbols for the Description of Conformations of Polynucleotide Chains. IUPAC-IUB Joint Commission on Biochemical Nomenclature (JCBN): Recommendations 1982. www.chem.qmul.ac.uk/iupac/misc/pnuc1.html Tatyana Abramova; Review: Frontiers and Approaches to Chemical Synthesis of Oligodeoxyribonucleotides. Molecules 2013, 18, 1063-1075; doi:10.3390/molecules18011063. Avery, OT, MacLeod, CM and McCarty, M (1944). Studies on the chemical nature of the substance inducing transformation of Pneumococcal types. J. Exp. Med. 79, 137-158. Willi Bannwarth; Synthesis of Oligodeoxynucleotides by the Phosphite-Triester Method Using Dimer Units and Different Phosphorous-Protecting Groups. Helvetica Chimica Acta, Volume 68, Issue 7, pages 1907–1913, 13 November 1985. J.C. Biro, B. Benyó, C. Sansom, Á. Szlávecz, G. Fördös, T. Micsik, and Z. Benyó; A common periodic table of codons and amino acids. Biochemical and Biophysical Research Communications 306 (2003) 408–415. Marvin H. Caruthers; The Chemical Synthesis of DNA/RNA: Our Gift to Science. January 11, 2013 The Journal of Biological Chemistry, 288, 1420-1427. F Eckstein. Oligonucleotides and analogues: A practical approach. Edited by F Eckstein. Series editors: D. Rickwood and B.D. Hames. IRL PRESS. Oxford University Press. 1991. Froehler, B.C. Deoxynucleoside H-Phosphonate Diester Intermediates in the Synthesis of Internucleotide Phosphate Analogues Tetrahedron Lett. 1986, 27, 5575-5578. P. T. Gilham and H. G. Khorana; Studies on Polynucleotides. I. A New and General Method for the Chemical Synthesis of the C5″-C3″ Internucleotidic Linkage. Syntheses of Deoxyribo-dinucleotides. J. Am. Chem. Soc., 1958, 80, 6212–6222. Hall, R.H., Todd, A., and Webb, R.F. 1957. Nucleotides. Part XLI. Mixed anhydrides as intermediates in the synthesis of dinucleoside phosphates. J. Chem. Soc. 3291-3296. Jablonski, E.; Moomaw, E.W.; Tullis, R.H. & Ruth, J.L. Preparation of oligodeoxynucleotide-alkaline phosphatase conjugates and their use as hybridization probes. Nucl. Acid Res. 1986, 14, 6115-6128. Kabachnik, M.M.; Potapov, V.K.; Shabarova, Z.A. & Prokofiev, M.A. Dokl. Acad. Nauk SSSR 1971, 201, 858-861. H. G. Khorana, G. M. Tener, J. G.Moffatt and E. H. Pol, Chem. Ind., 1956, 1523. H. G. Khorana, W. E. Razzell, P. T. Gilham, G. M. Tener and E. H. Pol; SYNTHESES OF DIDEOXYRIBONUCLEOTIDES. J. Am. Chem. Soc., 1957, 79, 1002–1003. Y. Lapidot, H. G. Khorana; Studies on Polynucleotides. XXVIII. The Specific Synthesis of C3″-C5″-Linked Ribooligonucleotides (4). The Stepwise Synthesis of Uridylyl-(3″ → 5″)-adenylyl-(3″ → 5″)-uridylyl-(3″ → 5″)-uridine. J. Am. Chem. Soc., 1963, 85 (23), pp 3852–3857. Y. Lapidot, H. G. Khorana; Studies on Polynucleotides. XXIX. The Specific Synthesis of C3″-C5″-Linked Ribooligonucleotides(5). Homologous Adenine Oligonucleotides. J. Am. Chem. Soc., 1963, 85 (23), pp 3857–3862. Letsinger, R.L. & Mahadevan, V. J.; Oligonucleotide Synthesis on a Polymer Support. Am. Chem. Soc. 1965, 87, 3526-3527. Letsinger, R.L. & Mahadevan, V. J. ; Stepwise Synthesis of Oligodeoxyribonucleotides on an Insoluble Polymer Support. Am. Chem. Soc. 1966, 88, 5319-5324. McBride, L.J. & Caruthers, M.H. An investigation of several deoxynucleoside phosphoramidites useful for synthesizing deoxyoligonucleotides. Tetrahedron Lett. 1983, 24, 245-248. A. M. Michelson and Alexander R. Todd; Nucleotides part XXXII. Synthesis of a dithymidine dinucleotide containing a 3′: 5′-internucleotidic linkage. J. Chem. Soc., 1955, 2632-2638. DOI: 10.1039/JR9550002632. J. G. Moffatt, H. G. Khorana; D-XYLOSE-3-PHOSPHATE. J. Am. Chem. Soc., 1956, 78 (4), pp 883–884. Operation Manual. MilliGen/Biosearch Cyclone™ Plus DNA Synthesizer. Usman, N.; Ogilvie, K. K.; Jiang, M. Y.; Cedergren, R. J.; The automated chemical synthesis of long oligoribuncleotides using 2'-O-silylated ribonucleoside 3'-O-phosphoramidites on a controlled-pore glass support: synthesis of a 43-nucleotide sequence similar to the 3'-half molecule of an Escherichia coli formylmethionine tRNA. J. Amer. Chem. Soc. 1987, 109 (25): 7845–7854 D. H. Rammler , H. G. Khorana; Studies on Polynucleotides. XX.1 Amino Acid Acceptor Ribonucleic Acids (1). The Synthesis and Properties of 2″ (or 3″)-O-(DL-Phenylalanyl)-adenosine, 2″ (or 3″)-O-(DL-Phenylalanyl)-uridine and Related Compounds. J. Am. Chem. Soc., 1963, 85 (13), pp 1997–2002. Reese, C.B. & Tompson, E.A.; A new synthesis of 1-arylpiperidin-4-ols. J. Chem. Soc. Perkin Trans. I, 1988, 2881-2885. Roy, S. and Caruthers, M.; Synthesis of DNA/RNA and their analogs via phosphoramidite and H-phosphonate chemistries. Molecules 2013, 18, 14268-14284. The RNA World. Second Edition. Ed. R. F. Gesteland, T. R. Cech, J. F. Atkins. Cold Spring Harbor Laboratory Press. Cold Spring Harbor, New York. Monograph 37, 1999. Subhadeep Roy and Marvin Caruthers; Synthesis of DNA/RNA and Their Analogs via Phosphoramidite and H-Phosphonate Chemistries. Molecules 2013, 18, 14268-14284; doi:10.3390/molecules181114268. Scaringe, S.A.; Wincott, F.E. & Caruthers, M.H.; Preparation of 5′‐Silyl‐2′‐Orthoester Ribonucleosides for Use in Oligoribonucleotide Synthesis. J. Am. Chem. Soc. 1998, 120, 11820-11821. H. Schaller, G. Weimann , B. Lerch , H. G. Khorana; Studies on Polynucleotides. XXIV. The Stepwise Synthesis of Specific Deoxyribopolynucleotides (4). Protected Derivatives of Deoxyribonucleosides and New Syntheses of Deoxyribonucleoside-3″ Phosphates. J. Am. Chem. Soc., 1963, 85 (23), pp 3821–3827. H. Schaller, H. G. Khorana; Studies on Polynucleotides. XXV. The Stepwise Synthesis of Specific Deoxyribopolynucleotides (5). Further Studies on the Synthesis of Internucleotide Bond by the Carbodiimide Method. The Synthesis of Suitably Protected Dinucleotides as Intermediates in the Synthesis of Higher Oligonucleotides. J. Am. Chem. Soc., 1963, 85 (23), pp 3828–3835. H. Schaller, H. G. Khorana ; Studies on Polynucleotides. XXVII. The Stepwise Synthesis of Specific Deoxyribopolynucleotides (7). The Synthesis of Polynucleotides Containing Deoxycytidine and Deoxyguanosine in Specific Sequences and of Homologous Deoxycytidine Polynucleotides Terminating in Thymidine. J. Am. Chem. Soc., 1963, 85 (23), pp 3841–3851. Sergueev, D.S. & Shaw, B.R.; H-Phosphonate Approach for Solid-Phase Synthesis of Oligodeoxyribo-nucleoside Boranophosphates and Their Characterization. J. Am. Chem. Soc. 1998, 120, 9417-9427. Smith, M., Rammler, D.H., Goldberg, I.H. and Khorana, H.G. (1962). Studies on Polynucleotides. XIV. Specific Synthesis of the C3'-C5' Interribonucleotide Linkage. Synthesis of Uridylyl-(3'->5')-Uridine and Uridylyl-(3'->5')-Adenosine. J. Amer. Chem. Soc. 84, 430-440. Smith, M. and Khorana, H.G. (1958). Nucleoside polyphosphates. VI. An improved and general method for the synthesis of ribo-and deoxyribo-nucleoside-5' triphosphates. J. Amer. Chem. Soc. 80, 1141-1145. Sproat BS.; RNA synthesis using 2'-O-(tert-butyldimethylsilyl) protection. Methods Mol Biol. 2005;288:17-32. G. M. Tener, H. G. Khorana, R. Markham, E. H. Pol; Studies on Polynucleotides. II.1 The Synthesis and Characterization of Linear and Cyclic Thymidine Oligonucleotides. J. Am. Chem. Soc., 1958, 80 (23), pp 6212–6222. G. Weimann, H. Schaller , H. G. Khorana; Studies on Polynucleotides. XXVI. The Stepwise Synthesis of Specific Deoxyribopolynucleotides (6). The Synthesis of Thymidylyl-(3″ → 5″)-deoxyadenylyl-(3″ → 5″)-thymidylyl-(3″ → 5″)-thymidylyl-(3″ → 5″)-thymidine and of Polynucleotides Containing Thymidine and Deoxyadenosine in Alternating Sequence. J. Am. Chem. Soc., 1963, 85 (23), pp 3835–3841. Winnacker, Ernst L.; From genes to clones: introduction to gene technology. VCH, 1987. R. S. Wright, H. G. Khorana; Phosphorylated Sugars. I. A Synthesis of β-D-Ribofuranose 1-Phosphate. J. Am. Chem. Soc., 1956, 78 (4), pp 811–816. | 















 Figure 21: DNA/RNA synthesizer model 394.
Figure 21: DNA/RNA synthesizer model 394.




 According to Michael J. Meany phenotype emerges only from the interaction of gene and environment. The function of a gene cannot be separated from its cellular environment. Every trait results from the interaction of a gene with the environment. The development of defensive responses to a threat as a maternal effect is a well established theme in biology. Evidence for transgenerational, maternal effects has been published for two animal models – one for a plant and the other for an insect. If a plant gets provoked, say by getting eaten by an insect, it usually reacts with an increase in the synthesis of defensive molecules such as mustard oil glycosides in the case of the radish. These defenses defend against the next attacks. The seedlings derived from caterpillar-damaged radishes showed a significant change in glycosinolate profiles and altered trichrome expression. Since only the mother plants had been exposed to the caterpillars but not the seedlings, these changes have to be adaptive. There are more examples available such as the capacity for night flight in grasshoopers, the tail length of lizards, and the helmet size of water flies and many more are getting discovered the more scientists look for them. These are all determined by maternal effects acting through yet unknown mechanisms. In these examples, traits of the parents are transmitted to the offsprings in a nongenomic manner. The environmental experience of the mother is inherited through an epigenetic mechanism of inheritance causing a phenotypic variation in the offspring. Studies off rats looking for maternal effects revealed that naturally occurring variations in maternal care are associated with individual differences in hypothalamic-pituitary-adrenal (HPA) axis responses to stress. For example the adult offspring of a high licking and grooming mother showed increased hippocampal glucocorticoid receptor expression and enhanced glucocorticoid feedback sensitivity compared to the offspring of a low licking and grooming mother. The first offspring also showed a decrease in hypothalamic corticotrophin-releasing factor (CRF) expression and a more modest HPA response to stress. The result is that the adult offspring rats from frequently licking and grooming mothers are in general behaviorally less fearful and show more modest HPA responses to stress than the offsprings of low licking and grooming mothers. How is this accomplished? In vivo and in vitro studies suggest that the glucocorticoid receptor gene expression is altered through increased hippocampal serotonin (5-HT) activity at 5-HT7 receptors. The increased 5-HT activity results in an increase in the expression of the transcription factor, nerve growth-inducible factor A (NGFI-A) in the hippocampus. The non-coding exon 1 region of the hippocampal glucocorticoid receptor includes a promoter region, exon 17, containing a binding site for NGFI-A. Noncoding regions of the DNA do not code for functional gene products – proteins - and usually contain sequences that regulate the expression of the “downstream” coding segment. For example, exon 1 contains several promoter sequences that can alter gene expression. The exon 17 sequence functions as a promoter in neurons and is more active in the offspring of high licking and grooming mothers, suggesting that the use of this promoter is enhanced as a function of maternal care. Transcription factors such as nerve growth-inducible factor A regulate gene expression and provide a cellular interface between environment and gene. Many details of our behavior and appearance appear to be determined by gene regulation. A striking example of the power of gene regulation is seen in agouti mice (see figure 4), in which genetically identical twins can look entirely different in both color and size. For example, one mouse may be small and brown, but her twin sister may be obese and yellow. Another genetically identical sister may have a mottled look with both fur colors present but may fall in the middle of the weight range. The genome of each of these mice is the same, but the gene expression obviously differs. How can that be? Waterland and Jirtle in 2003 studied the influence of nutrition on adult metabolism in yellow agouti (Avy) mice and showed that research on epigenetic changes resulting from the environment can give clues into obesity in mice--and humans. Their results showed that dietary methyl supplementation of a/a female mice with extra folic acid, B12, choline, and betaine alter the phenotype of their Avy/a offspring via increased CpG methylation at the Avy locus. This locus is a transposable gene element. We conclude, the epigenome is what makes the difference.
According to Michael J. Meany phenotype emerges only from the interaction of gene and environment. The function of a gene cannot be separated from its cellular environment. Every trait results from the interaction of a gene with the environment. The development of defensive responses to a threat as a maternal effect is a well established theme in biology. Evidence for transgenerational, maternal effects has been published for two animal models – one for a plant and the other for an insect. If a plant gets provoked, say by getting eaten by an insect, it usually reacts with an increase in the synthesis of defensive molecules such as mustard oil glycosides in the case of the radish. These defenses defend against the next attacks. The seedlings derived from caterpillar-damaged radishes showed a significant change in glycosinolate profiles and altered trichrome expression. Since only the mother plants had been exposed to the caterpillars but not the seedlings, these changes have to be adaptive. There are more examples available such as the capacity for night flight in grasshoopers, the tail length of lizards, and the helmet size of water flies and many more are getting discovered the more scientists look for them. These are all determined by maternal effects acting through yet unknown mechanisms. In these examples, traits of the parents are transmitted to the offsprings in a nongenomic manner. The environmental experience of the mother is inherited through an epigenetic mechanism of inheritance causing a phenotypic variation in the offspring. Studies off rats looking for maternal effects revealed that naturally occurring variations in maternal care are associated with individual differences in hypothalamic-pituitary-adrenal (HPA) axis responses to stress. For example the adult offspring of a high licking and grooming mother showed increased hippocampal glucocorticoid receptor expression and enhanced glucocorticoid feedback sensitivity compared to the offspring of a low licking and grooming mother. The first offspring also showed a decrease in hypothalamic corticotrophin-releasing factor (CRF) expression and a more modest HPA response to stress. The result is that the adult offspring rats from frequently licking and grooming mothers are in general behaviorally less fearful and show more modest HPA responses to stress than the offsprings of low licking and grooming mothers. How is this accomplished? In vivo and in vitro studies suggest that the glucocorticoid receptor gene expression is altered through increased hippocampal serotonin (5-HT) activity at 5-HT7 receptors. The increased 5-HT activity results in an increase in the expression of the transcription factor, nerve growth-inducible factor A (NGFI-A) in the hippocampus. The non-coding exon 1 region of the hippocampal glucocorticoid receptor includes a promoter region, exon 17, containing a binding site for NGFI-A. Noncoding regions of the DNA do not code for functional gene products – proteins - and usually contain sequences that regulate the expression of the “downstream” coding segment. For example, exon 1 contains several promoter sequences that can alter gene expression. The exon 17 sequence functions as a promoter in neurons and is more active in the offspring of high licking and grooming mothers, suggesting that the use of this promoter is enhanced as a function of maternal care. Transcription factors such as nerve growth-inducible factor A regulate gene expression and provide a cellular interface between environment and gene. Many details of our behavior and appearance appear to be determined by gene regulation. A striking example of the power of gene regulation is seen in agouti mice (see figure 4), in which genetically identical twins can look entirely different in both color and size. For example, one mouse may be small and brown, but her twin sister may be obese and yellow. Another genetically identical sister may have a mottled look with both fur colors present but may fall in the middle of the weight range. The genome of each of these mice is the same, but the gene expression obviously differs. How can that be? Waterland and Jirtle in 2003 studied the influence of nutrition on adult metabolism in yellow agouti (Avy) mice and showed that research on epigenetic changes resulting from the environment can give clues into obesity in mice--and humans. Their results showed that dietary methyl supplementation of a/a female mice with extra folic acid, B12, choline, and betaine alter the phenotype of their Avy/a offspring via increased CpG methylation at the Avy locus. This locus is a transposable gene element. We conclude, the epigenome is what makes the difference.













.png)