Neurodegeneration and Neurodegenerative DiseasesWhat do we fear the most when we age? Most of us will most like fear the degeneration of our brain power the most. I admit that symptoms like forgetting names or places that I know I knew annoys me a lot. Therefore neurodegenerative disorders are some of the most feared illnesses in our society.
Alzheimer’s disease affects up to 10% of people over 65 years of age. This disease causes the progressive loss of memory and other mental faculties, leaving the individual confused and incompetent to care for him- or herself. Huntington’s disease is another relatively common neurodegenerative disorder. One in ten thousand individuals are affected. People inflicted with this disease make involuntary movements and become serverely emotionally disturbed as well as cognitively impaired. Prion diseases, including Creutzfeldt-Jakob disease, lead to mental and physical decline followed by death.
Many scientists are working and have been working to find a treatment or cure, still, no successful treatments are yet available for any of this diseases but some progress has been made during recent years. Scientific research in this field aims to understand the disease mechanism in order to develop successful treatments and to prevent the onset of symptoms in patients. Particular genetic traits appear to be linked to many of these diseases. For example, the chromosome and gene linked to Huntington’s disease, Freiderich’s ataxia, the prion genes linked to spongiform encephalopathies, as well as the triplet repeat mutations responsible for myotonic dystrophy have all been identified. Model systems in which to test potential therapies and prevention strategies have been developed and are employed to find a cure for this cruel and distressing diseases.
Familiar neurodegenerative disorders are Alzheimer’s, Parkinson’s, Huntington’s and Wilson’s disorders. Table 1 shows a list of autosomal neurodegenerative diseases including what is known about chromosomal linkage, genes, mutations and pathology.
Table 1. Autosomal dominant primary neurodegenerative diseases. Ch, chromosome; PrP, prion protein; T, tangles; LB, Lewy bodies; 1, is present or exists; AD, Alzheimer’s disease; PD, Parkinson’s disease; HD, Huntington’s disease; SOD, superoxide dismutase.
*SBMA is technically not autosomal dominant but it is probably dominant in its cellular mode of action. Alzheimer's disease (AD) is the most common form of dementia among older people which is a brain disorder that seriously affects a person's ability to carry out daily activities. AD begins slowly, it first involves the parts of the brain that control thought, memory and language. Over time, symptoms get worse. People may not recognize family members or have trouble speaking, reading or writing. They may forget how to brush their teeth or comb their hair. Later on, they may become anxious or aggressive, or wander away from home. Eventually, they need total care. The disease usually begins after age 60 and the risk of getting it goes up with age. The risk is also higher if a family member has had the disease. No treatment can yet stop the disease.
Amyotrophic lateral sclerosis (ALS) is a disease that attacks nerve cells called neurons in the brain and spinal cord. The job of neurons is to transmit messages from the brain and spinal cord to voluntary muscles - the ones that control arms and legs. At first, this causes mild muscle problems, some people notice trouble when walking or running, writing and may notice speech problems. Eventually they lose strength and cannot move. When the muscles in the chest fail they cannot breathe. A ventilator may help, but most people with ALS die from respiratory failure. The disease usually strikes between age 40 and 60. More men than women get it. No one knows what causes ALS. It can run in families, but usually it strikes at random. There is no cure. Medicines can relieve symptoms and, sometimes, prolong survival.
Friedreich's ataxia is an inherited disease that damages the nervous system. It affects the spinal cord and the nerves that control muscle movement in arms and legs. Symptoms usually begin between the ages of 5 and 15. The main symptom is ataxia, which is trouble coordinating movements. Specific symptoms include difficulty to walk, muscle weakness, speech problems, involuntary eye movements, scoliosis and heart palpitations. People with Friedreich's ataxia usually need a wheelchair 15 to 20 years after symptoms first appear. In severe cases, people become incapacitated. There is no cure.
Huntington's disease (HD) is an inherited disease that causes certain nerve cells in the brain to waste away. People are born with the defective gene, but symptoms usually don't appear until middle age. Early symptoms of HD may include uncontrolled movements, clumsiness or balance problems. Later, HD can take away the ability to walk, talk or swallow. Some people stop recognizing family members. Others are aware of their environment and are able to express emotions. If one of the parents has Huntington's disease, there is a 50-50 chance of getting it. A blood test can tell if the HD gene is present and if there is a risk to develop the disease. There is no cure. Medicines can help manage some of the symptoms, but cannot slow down or stop the disease.
Lewy body disease is one of the most common causes of dementia in the elderly that causes a loss of mental functions severe enough to affect normal activities and relationships. Lewy body disease happens when abnormal structures, called Lewy bodies, build up in areas of the brain. The disease may cause a wide range of symptoms, including changes in alertness and attention, hallucinations, problems with movement and posture, muscle stiffness, confusion and finally loss of memory. Lewy body disease can be hard to diagnose since it can be confused with Parkinson’s and Alzheimer’s disease. The disease usually begins between the ages of 50 and 85. The disease gets worse over time. There is no cure.
Parkinson's disease affects nerve cells, or neurons, in a part of the brain that controls muscle movement. In Parkinson's, neurons that make a chemical called dopamine die or do not work properly. Dopamine normally sends signals that help coordinate movements. Symptoms of Parkinson's disease may include trembling of hands, arms, legs, jaw and face, stiffness of the arms, legs and trunks, slowness of movement, poor balance and coordination. Parkinson's usually begins around age 60 but can start earlier. It is more common in men than in women. There is no cure for Parkinson's disease.
Spinal muscular atrophy (SMA) attacks nerve cells, called motor neurons, in the spinal cord. These neurons communicate with voluntary muscles. As neurons are lost muscles weaken. This can affect walking, crawling, breathing, swallowing and head and neck control. SMA runs in families. Parents usually have no symptoms, but still carry the gene. There are many types of SMA, and some of them are fatal. Life expectancy depends on the type of disease.
Spinocerebellar ataxia (SCA) a progressive and degenerative genetic disease containes multiple types, each of which could be considered a disease in its own right. The following is a list of some, not all, types of Spinocerebellar ataxia. The first ataxia gene was identified in 1993 for a dominantly inherited type. It was called “Spinocerebellar ataxia type 1" (SCA1). Subsequently, as additional dominant genes were found they were called SCA2, SCA3, etc. Usually, the "type" number of "SCA" refers to the order in which the gene was found. At this time, there are at least 29 different gene mutations which have been found. Many SCAs below fall under the category of polyglutamine diseases, which are caused when a disease-associated protein (i.e. ataxin-1, ataxin-3, etc.) contains a glutamine repeat beyond a certain threshold. In most dominant polyglutamine diseases, the glutamine repeat threshold is approximately 35, except for SCA3 which is beyond 50. Polyglutamine diseases are also known as "CAG Triplet Repeat Disorders" because CAG is the codon which codes for the amino acid glutamine. Many prefer to refer to these also as polyQ diseases since "Q" is the one-letter reference for glutamine.
Spinal and bulbar muscular atrophy (SBMA), also known as spinobulbar muscular atrophy, X-linked bulbo-spinal atrophy, X-linked spinal muscular atrophy type 1 (SMAX1) and Kennedy's disease (KD)— A X-linked, recessive, slow progressing, neurodegenerative disease associated with mutations of the androgen receptor (AR) gene resulting in the impairment of the AR that can be viewed as a variation of the disorders of the androgen insensitivity syndrome (AIS).
Dentatorubral-pallidoluysian atrophy (DRPLA) is an autosomal dominant pinocerebellar degeneration caused by an expansion of a CAG repeat encoding a polyglutamine tract in the atrophin-1 protein. It is also known as Haw River Syndrome and Naito-Oyanagi disease. Although this condition was perhaps first described by Smith et al. in 1958, and several sporadic cases have been reported from Western countries, this disorder seems to be very rare except in Japan.
There are at least eight neurodegenerative diseases that are caused by expanded CAG repeats encoding polyglutamine (polyQ) stretches. The expanded CAG repeats create an adverse gain-of-function mutation in the gene products. Of these diseases, DRPLA is most similar to Huntington disease.
John Hardy and Katrina Gwinn-Hardy Genetic Classification of Primary Neurodegenerative Disease. SCIENCE VOL 282 6 NOVEMBER 1998, 1075-1079. NIH: National Institute on Aging. NIH: National Institute of Neurological Disorders and Stroke. |
Neurodegeneration and Neurodegenerative Diseases
↧
↧
Amino Acid Analysis
Amino Acid Analysis0. IntroductionThe analysis of free amino acids present in food samples, body fluids such as urine, serum and blood, and other sources, amino acid hydrolysates, from proteins, or primary and secondary amines is an important, standardized method routinely performed in biochemical, medical and biological labs. It has been, and still is used for the accurate quantification and characterization of proteins and peptides, as well as recombinant gene products. It is considered the method of choice to determine the purity and chemical composition of a protein or peptide. • A single hydrolysis can not yield quantitative recoveries for all amino acids present in the sample. • Losses of up to 50-100% can be experienced for some amino acids. • Adding reductants and/or scavengers to the hydrolysis acid will improve yields of amino acids sensitive to hydrolysis conditions (ser, thr, met, tyr). • Shorter hydrolysis times and/or lower hydrolysis temperatures will improve yields of amino acids sensitive to hydrolysis conditions but may obscure others. 3b. Hydrolysis of sensitive amino acids • Serine and threonine
Side chain hydroxyl group is modified during hydrolysis (eg. esterification, dehydration). Typical losses using standard hydrolysis conditions are 15-20% for serine and 10-15% for threonine. A typical method for quantitation is to run multiple hydrolyses at different hydrolysis times and plot the serine and threonine recovery versus length of hydrolysis (hydrolyzed for 30, 60 and 90 min). Extrapolate the recovery to time = 0 to yield an accurate quantitation. • Tyrosine Typical losses are 15-20% during hydrolysis (actual losses may be higher depending on the quality of acid used and sample amount). Side chain phenol group is attacked by traces of hypochlorite/chlorine free radicals present in HCl. Addition of scavengers is necessary to protect tyrosine. Typically phenol is added to the acid (0.1 to 1% by weight). The quality of the phenol is important. Poor quality phenol will not protect tyrosine. • Methionine Losses of methionine can vary depending on sample amount, quality of HCl, amount of oxygen present in the hydrolysis vessel, length of time the hydrolyzed sample is exposed to air on the sample slide etc. Side chain thioether is oxidized forming methionine sulfone and sulfoxide. Addition of a reductant/scavenger improves methionine yields. Choice of reductants needs to be done carefully. Some reductants (thioglycolic acid, DTT, 2-mercapto-ethanol) react with PITC through their free sulfhydryl group and generate peaks that can interfere with the PTC-amino acid analysis. Borane-DIEA should not be used with the hydrolyzer. • Cysteine/Cystine Losses can be 50% or greater. The free sulfhydryl and disulfide groups are sensitive to a variety of side reactions during hydrolysis. It is necessary to reduce the disulfide bonds and alkylate or oxidize the free sulfhydryl groups generated. Derivatization to form pyridylethyl or carboxy-methyl cysteine or oxidation to cysteic acid are typical techniques to quantitate this amino acid. One recommended technique is derivatization using 4-vinyl pyridine to form pyridylethyl cysteine. • Tryptophan Mostly destroyed during hydrolysis by attack on the carbon double bond in the indole ring. It is not possible to get quantitative recovery of tryptophan from samples using acid hydrolysis. In manual hydrolysis the addition of thioglycolic acid to the acid (5-15% by volume) has given 70% recovery of trp at 500 pmol. Thioglycolic acid does generate a large interfering artifact peak in the PTC-chemistry. A method using dodecanthiol as a scavenger has proven to be useful. • Asparagine and Glutamine Quantitatively recovered as aspartic and glutamic acid respectively. Many attempts have been made over the years to improve recoveries of hydrolysis sensitive amino acids as well as to generate overall quantitative recoveries for all studied amino acids. A collection of hydrolysis conditions studied is listed in table 1. Table 1: Protein/Peptide Hydrolysis Methods
4. Derivatization methods for the amino acid analysis of proteins and peptides Many pre-column derivatization chemistries have been investigated over the years. All of them suffer from major disadvantages such as incompatibility with aqueous samples or dissolved salts, or interference from reagent peaks in the analysis chromatogram. A list of derivatization methods commonly used is shown in the next table: Table 2: Derivatization Chemistries for Amino Acids and Amine Analysis
A "pre-column" derivatization method used by several companies which produce and marked amino acid analyzer typically consists of several steps as listed below: 1. Derivatization of amino acids 2. Separation of derivatized amino acids by reversed phase chromatography 3. Detection in UV or using fluorescence for increased sensitivity (some chemistries). A "post-column" derivatization method mainly used for on-line ninhydrin derivatization in an automated amino acid analyzer consists of several steps as listed below: 1. Separation of amino acids by ion exchange chromatography 2. Derivatization of amino acids with ninhydrin at an elevated temperature 3. Detection of derivatives via absorption in the visible range (440 and 570 nm). The ninhydrin based system has been the most widely used system. 4.1. Derivatization of free amino acids using phenylisothiocyanate (PITC) 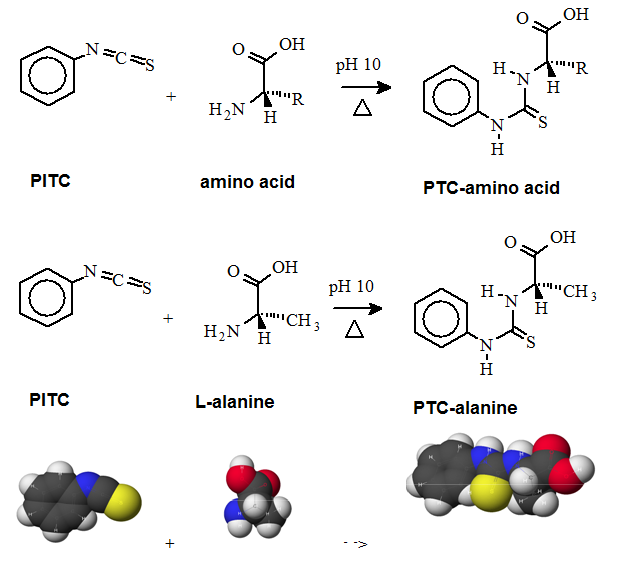 Figure 1: Derivatization reaction of amines and free amino acids using PITC. PITC reacts with the free amino groups in amines and amino acids to form the phenylthiourea adduct of these compounds making them suitable for UV-detection.This reaction is used in the automated hydrolyzer/derivatizer set-up with on-line HPLC separation of the resulting PTC-amino acids and UV detection. Detection is done at 268 to 270 nm. 4.2 Derivatization of free amino acids using ortho-phthalaldehyde (OPA)  Figure 2: Derivatization reaction of amines and free amino acids using ortho-phthalaldehyde (OPA). OPA reacts with the free amino groups in aminesand amino acids in the presents of a reducing reagent like b-mercaptoethanol to form their isoindole-derivatieves making them suitable for UV-and fluorescence detection.This reaction is usually used in a pre-column derivatization step with an automated derivatizer set-up with on-line HPLC separation. UV detection is done at 338 nm. Fluorescence detection is done using excitation settings at 340 nm and emmission settings at 450 nm. 4.3. Derivatization of free amino acids using (FMOC-Cl) 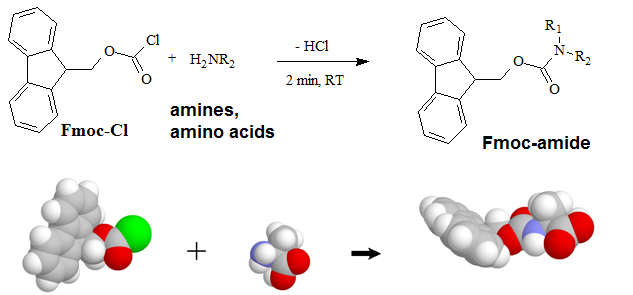 Figure 3: Derivatization reaction of amines and free amino acids using 9-fluorenylmethyl-chloroformat (Fmoc). Fmoc reacts with the free amino groups in amines and amino acids to form Fmoc-derivatieves making them suitable for UV-and fluorescence detection.This reaction is usually used in a pre-column derivatization step with an automated derivatizer set-up with on-line HPLC separation. UV detection is done at 262 nm. Fluorescence detection is done using excitation settings at 266 nm and emmission settings at 305 nm. 4.4. Derivatization using Waters AccQ•TagTM amino acid analysis system  Figure 4A: AQC (6-aminoquinolyl-N-hydroxy-succinimidyl carbamate. Chemical structure (left) and energy minimized molecular model (right). Calculations were done using the MNDO module from CACHE Scientific. 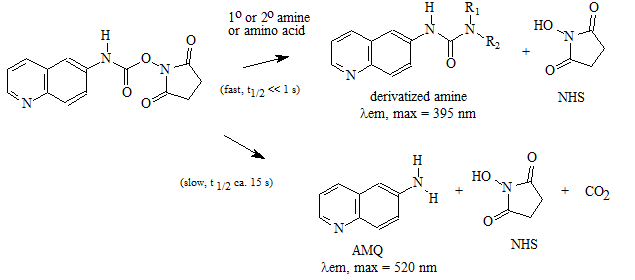 Figure 4B: Derivatization Chemistry. Both 1º and 2° amino acids and amines react rapidly with AQC to produce highly stable, fluorescent derivatives. The excess reagent reacts with water to form a free amine having significantly different fluorescence spectral properties. ReferencesBarkholt, V. and Jensen, A., Anal. Biochem., (1989) 177, 318-322. Betner, I./ Foldi, P. New Automated Amino Acid Analysis by HPLC Precolumns Derivatization with Fluorenylmethyloxycarbonylchloride. Chromatographia Vol. 22, No. 7-12, Dec. 1986 -OPA/FMOC Capony and Demaille, Anal. Biochem. 152, 206-212 (1983). Carlson, R./ Srinivasachar, K./ Givens, R./ Matuszewski, B. New Derivatizing Agents fore Amino Acids and Peptides. 1. Facile Synthesis of N-Substituted 1-Cyanobenz[f]isoindoles and their Spectroscopic Properties. American Chemical Society (1986) 51, pg. 3978. -NDA Chan, King/ Janini, George/ Muschik, Gary/ Issaq, Haleem. Laser-induced fluorescence detectin of 9-fluorenylmethyl chloroformate derivatized amino acids in capillary electrophoresis. Journal of Chromatography A, 653 (1993) 93-97 -OPA/FMOC-Cl/NDA Cohen, Phillip/ Hubbard, Michael. On target with a new mechanism for the regulation of protein phosphorylation. TIBS 18, May 1993 pgs. 172-177. Cohen, S. and Michaud, D., Anal. Biochem., (1993) Cohen, s., and Strydom, D., Anal. Biochem., (1988) 174, 1-16. Cohen, Steven/ Tarvin, Thomas/ Bidlingmeyer, Brian.Analysis of amino acids using precolumn derivatization with phenylisothiocyanate.American Laboratory Aug 1984. -PITC D'Aniello, Antimo/ Petrucelli, Leonard/ Gardner, Christina/ Fisher, George. Improved Method for Hydrolyzing Proteins and Peptides without Inducing Racemization and for Determining Their True D-Amino Acid Content. Analytical Biochemistry 213, 290-295 (1993) Hancock, Diane/ Reeder, Dennis. Analysis and configuration assignments of the amino acids in a pyroverdine-type siderophore by reversed-phase high-performance liquid chromatography. Journal of Chromatography, 646 (1993) 335-343. -PITC Hariharan, M./ Naga, Sundar/ VanNoord, Ted. Systematic approach to the development of plasma amino acid analysis by high-performance liquid chromatography with ultraviolet detection with precolumn derivatization using phenyl isothiocyanate. Journal of Chromatography, 621 (1993) pgs. 15-22. -PITC Janssen, P./ van Nispen,J./ Melgers, P./ van den Bogaart, H./ Hamelinck, R./ Goverde, B. HPLC Analysis of Phenylthiocarbamyl (PTC) Amino Acids. I. Application in the Analysis of (Poly)peptides. Chromatographia Vol. 22, No. &-12, pg. 351-357, Dec. 1986. -PITC Jonge, Leon/ Breuer, Michel. Modification of the analysis of amino acids in pig plasma. Journal of Chromatography B, 6542 (1994) 90-96. Kawasaki , Takao/ Higuchi, Takeru/ Imai, Kazuhiro/ Wong, Osborne. Determination of Dopamine, Norepinephrine, and Related Trace Amines by Prochromatographic Derivatization with Naphthalene-2,3-dicarboxaldehyde. Analytical Biochemistry 180, 279-285 (1989) -NDA-CN Kemp, B.E. (1980) Relative alkali stability of some peptide o-phosphoserine and o-phosphothreonine esters. FEBS Lett.110, 308-312. LeFevre, Joseph. Reversed-phase thin-layer chromatographic separations of enantiomers of dansyl-amino acids using B-cyclodextrin as a moblie phase additive. Journal of Chromatography A, 653 (1993) 293-302 Liu, T.-Y. and Chang, Y.H. (1971) Hydrolysis of proteins with p-toluenesulphonic acid. Determination of tryptophan. J. Biol. Chem. 246, 2842-2848. Lobell, Mario/ Schneider, Manfred. 2,3,4,6-Tetra-O-benzoyl-B-D-glucopyranosyl isothiocyanate:an efficient reagent for the determination of enantiomeric purities of amino acids, B-adrenergic blockers and alkyloxiranes by high-performance liquid chromatography using standard reversed-phase columns. Journal of Chromatography, 633 (1993) 287-294. AGIT Lunte, Susan. Naphthalenedialdehyde-cyanide:A versatile fluorogenic reagent for the LC analysis of peptides and other primary amines. >LC-GC Vol. 7, No. 11 pg. 908-916 -NDA-CN Lunte, Susan/ Mohabbat, Tariq/ Wong, Osborne/ Kuwana, Theodore. Determinatin of Desmosine Idodesmosine, and Other Amino Acids by Liquid Chromatography with Electrochemical Detection following Precolumn Derivatization with Naphthalenedialedhyde/Cyanide. Analytical Biochemistry 178, 202-207 (1989) -NDA Martensen, T.M. (1982) Phosphotyrosine in proteins. Stability and quantification. J. Biol. Chem. 257, 9648-9652. Matsubara & Sasaki, BBRC 35, 175-181 (1969). Matuszewski, Bogdan/ Givens, Richards/ Srinivasachar, Kasturi/ Carlson, Robert/ Higuchi, Takeru. N-Substituted 1-Cyanobenz[f]isoindole: Evaluation of Fluoresence Efficiencies of a New Fluorogenic Label for Primary Amines and Amino Acids. Analytical Chemistry, Vol. 59, Page 1102, (1987) -NDA-CN Montigny, Pierre/ Stobaugh, John/ Givens, Richard/ Carlson, Robert/ Srinivasachar, Kasturi/ Sternson, Larry/ Higuchi, Takeru. Naphthalene-2,3-dicarboxaldehyde/Cyanide Ion: A Rationally Designed Fluorogenic Reagent for Primary Amines. Analytical Chemistry (1987) Vol. 59, pg. 1096. -NDA-CN Moore, S. and Stein, W.H. (1951) J. Biol. Chem. 178, 53-77. Moore, S. and Stein, W.H. (1954) J. Biol. Chem. 211, 893-906. Moore, S. and Stein, W.H. (1963) Methods Enzymol. 6, 819-831 (1963). Neidle, Amos/ Banay-Schwartz, Miriam/ Sacks, Shirley/ Dunlop, David. Amino Acid Analysis Using 1-Napthylisocyanate as a Precolumn High Performance Liquid Chroamatography Derivatization Reagent. Analytical Biochemistry 189 (1989) 291-297. -PITC/ OPA/ FMOC-Cl Nimura, N., Iwaki, K., Kinoshita, T., Takeda, K. and Ogura, H. (1986) Anal. Chem. 58, 2372-2375. Organon, Janssen. PTC-Amino Acid separation: C18 column. Chromatographia, 22: 345-357 (1986) -PTC Penke et al., Anal. Biochem. 60, 45-50 (1974). Roach, Marc/ Harmony, Marlin. Determination of Amino Acids at Subfemtomole Levels by High-Performance Liquid Chromatography with Laser-Induced Fluorescence Detection. Analytical Chemistry, 1987, vol. 59 pg. 411. -OPA Slater, George/ Manville, John. Analysis of thiocyanates and isothiocyanates by ammonia chemical ionization gas chromatography-mass spectrometry and gas chromatography-Fourier transform infrared spectroscopy. Journal of Chromatography, 648 (1993) 433-443 -ITC Strydom, D., and Cohen, S., in Techniques in Protein Chemistry IV (R.H. Angeletti, ed.), Academic Press, (1993) San Diego Strydom, D., Tarr, G., Pan, Y-C and Paxton, R., in Techniques in Protein Chemistry II (R.H. Angeletti, ed.), Academic Press, (1992) San Diego , pp. 261-274. Swadesh, J.K., Thannhauser, T.W., an Scheraga, H.A. (1984) Sodium sulphite as an antioxidant in the acid hydrolysis of bovine pancreatic ribonuclease A. Anal. Biochem. 171, 133-123, 397-401 ibid? Tsugita & Scheffler, Eur. J. Biochem. 124, 585-588 (1982). vanEijk, Hans/ Rooyakkers, Dennis/ Deutz, Nicolaas. Rapid routine determination of amino acids in plasma by high performance liquid chromatography with a 2-3 mm Spherisorb ODS II column. Journal of Chromatography, 620 (1993) 143-148. - OPA Waldron, Karen/ Dovichi, Norman. Sub-Femtomole Determination of Phenylthiohydantion-Amino Acids: Capillary Electrophoresis and Thermooptical Detection. Anal. Chem. (1992) 64, 1396-1399 -? Westall & Hesser, Anal. Biochem. 61, 610-613 (1974). Woo, Kang-Lyung/ Chang, Duk-Kyu. Determination of 22 protein amino acids as N(O)-tert-butyldimethylsiyl derivatives by gas chromatography. Journal of Chromatography, 638 (1993) 97-107. Wu, Guoyao. Determination of proline by reversed-phase high-performance liquid chromatography with automated pre-column o-phthaldialdehyde. Journal of Chromatography, 641 (1993) 168-175 -OPA Yokote, et al., Anal. Biochem. 152, 245-249 (1986). Zhou, F./ Krull, I. Solid-phase derivatization of amino acids and peptides in high-performance liquid chromatography. Journal of Chromatography, 648 (1993) 357-365. -FMOC-Cl. |
↧
Abbreviation for Antibody Species Reactivity
<div><b><font size="4">Abbreviations</font></b></div> <table> <tbody> <tr> <td width="50">Ar</td> <td>Arabidopsis</td> </tr> <tr> <td>B</td> <td>Bovine</td> </tr> <tr> <td>BA</td> <td>Baboon</td> </tr> <tr> <td>Bc</td> <td>Bacteria</td> </tr> <tr> <td>Bl</td> <td>Blocking</td> </tr> <tr> <td>Bt</td> <td>Biotinylated</td> </tr> <tr> <td>Ce</td> <td>C. elegans</td> </tr> <tr> <td>C</td> <td>Chicken</td> </tr> <tr> <td>Chm</td> <td>Chimpanzee</td> </tr> <tr> <td>CP</td> <td>Carp</td> </tr> <tr> <td>CT</td> <td>C-Terminus</td> </tr> <tr> <td>D</td> <td>Dog</td> </tr> <tr> <td>Ds</td> <td>Drosophila</td> </tr> <tr> <td>E</td> <td>ELISA</td> </tr> <tr> <td>ED</td> <td>Extracellular Domain</td> </tr> <tr> <td>EL</td> <td>Extracellular Loop</td> </tr> <tr> <td>F</td> <td>Feline</td> </tr> <tr> <td>FACS</td> <td>Flow Cytometry</td> </tr> <tr> <td>Fu</td> <td>Fungus</td> </tr> <tr> <td>G</td> <td>Guinea Pig</td> </tr> <tr> <td>Gt</td> <td>Goat</td> </tr> <tr> <td>H</td> <td>Human</td> </tr> <tr> <td>HM</td> <td>Hamster</td> </tr> <tr> <td>Hr</td> <td>Horse</td> </tr> <tr> <td>I</td> <td>Immunogen</td> </tr> <tr> <td>ICC</td> <td>Immunocytochemistry</td> </tr> <tr> <td>ID</td> <td>Intracellular Domain</td> </tr> <tr> <td>IF</td> <td>Immunofluorescence</td> </tr> <tr> <td>IHC</td> <td>Immunohistochemistry</td> </tr> <tr> <td>IN</td> <td>Intermediate Domain</td> </tr> <tr> <td>IP</td> <td>Immunoprecipitation</td> </tr> <tr> <td>M</td> <td>Mouse</td> </tr> <tr> <td>Ma</td> <td>Mammal</td> </tr> <tr> <td>Mk</td> <td>Monkey</td> </tr> <tr> <td>MS</td> <td>Mass Spectrometry</td> </tr> <tr> <td>Mt</td> <td>M. tuberculosis</td> </tr> <tr> <td>N</td> <td>Neutralization</td> </tr> <tr> <td>NT</td> <td>N-Terminus</td> </tr> <tr> <td>P</td> <td>Porcine</td> </tr> <tr> <td>Pa</td> <td>Parasite</td> </tr> <tr> <td>PF</td> <td>Puffer Fish</td> </tr> <tr> <td>Pl</td> <td>Plant</td> </tr> <tr> <td>Pz</td> <td>Protozoan</td> </tr> <tr> <td>R</td> <td>Rat</td> </tr> <tr> <td>Rb</td> <td>Rabbit</td> </tr> <tr> <td>S</td> <td>Sheep</td> </tr> <tr> <td>V</td> <td>Virus</td> </tr> <tr> <td>WB</td> <td>Western Blot</td> </tr> <tr> <td>X</td> <td>Xenopus</td> </tr> <tr> <td>Y</td> <td>Yeast</td> </tr> <tr> <td>ZF</td> <td>Zebra Fish</td> </tr> </tbody> </table> <br />
↧
Abbreviations for Antibody Tested Applications
Abbreviations
| Ar | Arabidopsis |
| B | Bovine |
| BA | Baboon |
| Bc | Bacteria |
| Bl | Blocking |
| Bt | Biotinylated |
| Ce | C. elegans |
| C | Chicken |
| Chm | Chimpanzee |
| CP | Carp |
| CT | C-Terminus |
| D | Dog |
| Ds | Drosophila |
| E | ELISA |
| ED | Extracellular Domain |
| EL | Extracellular Loop |
| F | Feline |
| FACS | Flow Cytometry |
| Fu | Fungus |
| G | Guinea Pig |
| Gt | Goat |
| H | Human |
| HM | Hamster |
| Hr | Horse |
| I | Immunogen |
| ICC | Immunocytochemistry |
| ID | Intracellular Domain |
| IF | Immunofluorescence |
| IHC | Immunohistochemistry |
| IN | Intermediate Domain |
| IP | Immunoprecipitation |
| M | Mouse |
| Ma | Mammal |
| Mk | Monkey |
| MS | Mass Spectrometry |
| Mt | M. tuberculosis |
| N | Neutralization |
| NT | N-Terminus |
| P | Porcine |
| Pa | Parasite |
| PF | Puffer Fish |
| Pl | Plant |
| Pz | Protozoan |
| R | Rat |
| Rb | Rabbit |
| S | Sheep |
| V | Virus |
| WB | Western Blot |
| X | Xenopus |
| Y | Yeast |
| ZF | Zebra Fish |
↧
Bridged nucleic Acids (BNA) - BNA3
Bridged Nucleic Acids (BNAs) - BNA3Beyond peptide nucleic acids (PNAs) and locked nucleic acids (LNAs)The near completion of the human genome sequence in 2001 as well as the availability of whole genome sequences for many organisms in recent years has led to an exponential increase in genomic information in the last decade. The NIH website, http://www.ncbi.nlm.nih.gov/genome, contains a collection of sequences for partial or whole genomes, the number of which is constantly increasing at a high speed. This huge amount of sequence information provides a large source for the design of genetic tools to study molecular details of the genetic information flow in organisms. Scientific evidence shows that humans and other vertebrates are made up by close to or more than 220 different specialized cell types. Each cell type has its own gene regulatory network. To understand an organism at the molecular level the regulation of the genetic information flow will need to be studied. For this purpose new tools have been and are constantly developed. The double helix of DNA is nature’s solution to storing, retrieving, and communicating genetic information of a living organism. Two of many important characteristics of the DNA molecule are the specificity and the reversible nature of the hydrogen bonding between complementary nucleobases. These properties allow the DNA strands of the double helix to unwind and rewind in exactly the same configuration. It is now understood that the cell contains a vast collection of ‘nano-machines’ that control gene regulation. DNA, RNA, proteins, and other molecules make up these nano-assemblies. Control points for gene expression that are studied by scientists include transcriptional regulation, RNA processing, translational control, the stability of mRNA, posttranslational control, and DNA rearrangements among others. These regulatory systems found in prokaryotes and eukaryotes can differ from each other in many details. Once the nature of the DNA and RNA molecules was established the field of life science realized early on that if specific, single strands of DNA could be synthesized, scientist would be able to study and manipulate the gene sequences of DNA and RNA. This was realized with the development of efficient chemistries including automated instrumentation to allow for the synthesis of DNA and RNA monomers and polymers of increased size including their modifications. Oligonucleotide chemistry has been developed greatly over the past three decades and many advances have been made in the design of DNA, RNA and peptide based molecular tools with increased nuclease resistance.
The double helix exists in multiple conformations which were revealed by X-ray diffraction studies of concentrated DNA solutions that had been drawn out into thin fibers. Two kinds of structures, the B and the A forms of DNA were found. It is thought that the B form corresponds to the average structure of DNA under physiological conditions. Its structure has 10 base pairs per turn, and a wide major groove and a narrow minor groove. The A form on the other hand has 11 base pairs per turn and its major groove is narrower and much deeper than that of the B form. Furthermore, its minor groove is broader and shallower. The B DNA is found in the vast majority of the DNA in the cell. However, A DNA can be found in certain DNA-protein complexes. The non-covalent bonds in double stranded DNA (dsDNA) sequences are formed by Watson-Crick interactions. On the other hand, in triplex forming oligonucleotides the triplex forming strand binds to the duplex via Hoogsteen hydrogen interactions. Structures of the nucleotides and their interaction are shown in the following figure.
 Synthetic oligonucleotides are now important, established tools for life scientists and have many applications in molecular biology, genetic diagnostics and are poised to become important tools in the emerging field of molecular medicine as well. While unmodified oligodeoxynucleotides can form DNA:DNA and DNA:RNA duplexes they are sometimes unstable and labile to nucleases. Therefore a variety of nucleic acid analogs have been developed to enhance high-affinity recognition of DNA and RNA targets, enhancing duplex stability and assist with cellular uptake. Among the list of these analogs are peptide nucleic acids (PNAs), 2’-fluoro N3-P5-phosphoamidites, 1’, 5’-anhydrohexitol nucleic acids (HNAs) and locked nucleic acids (LNAs) as well as other bridged nucleic acids (BNAs). Peptide nucleic acids (PNAs) are synthetic polymers that contain a peptide backbone and nucleic acid bases as side chains within their sequence. These polymers can form strong specific bonds with complementary sequences present in double-stranded DNA (dsDNA). 2’-fluoro N3-P5-phosphoamidites are modified nucleotide analogs that contain fluor in the 2’ position. Hexitol nucleic acids are oligonucleotides built up from natural nucleobases and a phosphorylated 1,5-anhydrohexitol backbone. Anhydrohexitol oligonucleotides can be synthesized using phosphoramidite chemistry and standard protecting groups. LNAs are structurally rigid oligo-nucleotides with increased binding affinities.
A bridged nucleic acid (BNA) is a molecule that can contain a five-membered or six-membered bridged structure. Ideally the bridge is synthetically incorporated at the 2’, 4’-position of the ribose to afford a 2’, 4’-BNA monomer. The monomers can be incorporated into the oligonucleotide polymer structure using standard phosphoamidite chemistry. The goal for the synthesis and use of BNAs is to generate oligonucleotides with (i) equal or higher binding affinity against an RNA complement with excellent single-mismatch discriminating power, (ii) much better RNA selective binding, (iii) stronger and more sequence selective triplex-forming characters, and (iv) with a pronounced higher nuclease resistance, even higher than Sp-phosphorthioate analogues, than regular DNA or RNA oligonucleotides.
While a large number of chemically modified oligonucleotides have been developed during the last few decades, most of these molecules have failed to give the desired response and the search for new molecules with better qualities still continues today. One dramatic improvement was made with the introduction of the bridged nucleic acid, 2’, 4’-BNA (also called LNA). The compound shows a better hybridization affinity for complementary strands, both for RNA and DNA strands, in comparison to unmodified nucleotides. Furthermore the BNA can be used to design sequence selective LNA-oligonucleotide hybrids that are soluble in aqueous solutions and exhibit improved biostability in comparison to natural nucleotides. This BNA monomer has now been widely used in nucleic-acid-based technologies. However, according to Imanishi’s group, there is a need for further development because the nuclease resistance of the LNA is significantly lower than that obtained by phosphorothioate oligonucleotide and the fact that oligonucleotides containing consecutive LNA units or a fully modified oligonucleotide using the analog are very rigid and inflexible. Furthermore, additional research has proven that a kind of LNA-modified antisense oligonucleotide is hepatotoxic.
The scientists Satoshi Obika, Daishu Nanbu, Yoshiyuki Hari, Ken-ichiro Morio, Yasuko In, Toshimasa Ishida, and Takeshi Imanishi in 1997 reported the synthesis of a 2’-O,4’-C-Methyleneneuridine and –cytidine, novel bicylic nucleosides with a fixed C3 - endo sugar puckering. One of these nucleoside analogs is now known as a “locked nucleotide” or LNA. In the ribofuranose, the plane C1’-O4’-C4’ is fixed. The C3’- endo conformation is found in RNA. DNA can adjust and is able to take on both conformations. The exact nomenclature of sugar puckers can be found at http://www.chem.qmul.ac.uk/iupac/misc/pnuc2.html
Obika et al. in 1998 (Imanishi’s research group) report the synthesis of bicyclic nucleoside analogues with a fixed N-type conformation, 2'-O,4'-C-methyleneuridine and –cytidine and the incorporated of this analogue into oligonucleotides. The binding efficiency of the modified oligonucleotides to the complementary DNA and RNA as well as the CD spectra of the modified DNA-DNA and modified DNA-RNA duplexes were studied.
Imanishi’s group has synthesized newer generations of BNAs with improved properties, one called 2’4’-BNA-COC, and more recently, one called 2’,4’-BNA-NC. To optimize the length of the bridging moiety the BNA-NC was designed to contain a six-membered bridged structure with an N-O bond in the sugar molecule. The next figures show the chemical structures and 3D ball-and-stick model for the three BNAs.
Hari et al. in 2003 developed a novel nucleoside analogue for the effective recognition of CG interruption in a homopurine–homopyrimidine tract of double-stranded DNA (dsDNA). The group succeeded to synthesize a triplex-forming oligonucleotide (TFO) containing a novel 2’,4’-BNA (QB) bearing 1-isoquinolone as a nucleobase. The group used this BNA to investigate the triplex-forming ability and sequence-selectivity of the TFO. Using melting temperature (TM) measurements, it was found that the TFO-QB formed a stable triplex DNA in a highly sequence-selective manner under near physiological conditions.
Rahman et al. reported in 2007 that oligonucleotides containing 2’, 4’-BNA-NC monomers show an increase in their melting temperature (Tm) of 5.3 to 6.3 °C per modification (ΔT/mod) when investigated by UV melting experiments (Tm measurements).The melting temperature (Tm) is defined as the temperature at which half of the DNA strands are in the double-helical state and half are in the random coil state. The Tm depends on the length of the oligonucleotide molecule and its sequence. The Tm values of duplexes formed by 2’, 4’-BNA-NC[NH] oligonucleotides that contained three or more modifications are higher than those exhibited by the corresponding 2’, 4’-LNA modified oligonucleotides indicating a stronger binding and stability of the duplexes. Both 2’, 4’-BNA-NC[NH] and 2’, 4’-BNA-NC[NCH3] display selective binding to RNA, superior to that of 2’, 4’-LNAs. The researchers used mismatch discrimination studies to evaluate their ability for selective hybridization. The presence of a mismatched base in the target RNA strand resulted in a substantial decrease in the Tm of the duplexes formed. The analysis of formed duplexes by circular dichroism (CD) verified the presence of the A-form by showing the spectra typical for this structure. The A-form of the helical structure is favored because of the conformational restriction of the sugar moiety to the N-form. Furthermore, the BNAs can form triplexes with a higher stability compared to that of LNAs. Applications in antigenic and gene repair technologies require the formation of stable triplexes at physiological pH. Next, to test the BNAs for their resistance to nucleases oligonucleotides [5’-d(TTTTTTTTTT)-3’] modified with a single 2, 4’-BNA-NC unit were incubated with a 3’-exonuclease (Crotalus adamanteus venom phosphodiesterase, CAVP, Pharmacia) at 37 °C and the amount of intact oligonucleotides remaining were evaluated by RP-HPLC. The results showed that oligonucleotides modified with BNA-NC[NMe] are much more resistant to degradation than LNA modified oligonucleotides.
Rahman et al. in 2007 report on the use of BNA analogues to allow the formation of highly stable pyrimidine-motif triplexes at physiological pH. The formation of a stable triplex DNA molecule at physiological pH values is a highly desirable phenomenon in molecular biology and medicinal chemistry because of its function in the regulation of gene expression, site-specific cleavage of DNA, gene mapping and isolation, the maintenance of folded chromosome conformations, and gene-targeted mutagenesis. The researchers argue that in a pyrimidine-motif triplex DNA, the (homopyrimidine) triplex-forming oligonucleotide (TFO) binds to the homopurine tract of the target duplex DNA. This binding is sequence specific and maintained through Hoogsteen hydrogen bonds to form T·A:T and C+·G:C triads. The formation of the C+·G:C triad depends on the protonation of cytosine, which is only favorable at acidic pH values (pKa=4.5). Therefore, homo-pyrimidine-motif triplexes are extremely unstable at physiological pH values severely limiting their biological application. The group synthesized a novel bridged nucleic acid analogue, 2’,4’-BNANC, and demonstrated that the TFOs composed of 2’,4’-BNANC formed highly stable pyrimidine-motif triplexes at physiological pH values. Furthermore they show that the TFOs eliminate the requirement of placing alternating DNA monomers for optimum efficacy needed when using LNAs. They could show that fully modified TFOs still formed a highly stable triplex. They stated that “these promising properties of 2’,4’-BNANC will be helpful for developing oligonucleotide-based technologies for the postgenome era.”
Also in 2007, Imanishi’s group synthesized oligonucleotides that were modified with a novel BNA analogue, 2’, 4’-BNANC[N–CH3], and compared their properties to oligonucleotides containing 2’,4’-BNA (LNA). They showed that these oligonucleotides have a similarly high RNA affinity but a better selectivity for RNA and a much higher resistance to nuclease degradation. These results suggested that the novel BNA analogue may be particularly useful for antisense approaches when used for the design of antisense oligonucleotides.
Kasahara et al. in 2010 published a paper in where they showed that the capping of the 3’-ends of thrombin binding aptamers (TBAs) with bridged nucleotides increased the nuclease resistances and the stability of the aptamers in human serum. The capping did not affect the binding abilities of the aptamers. The researchers report that the capping could be achieved via a one step enzymatic process using 2’, 4’-bridged nucleoside 5’-triphosphate and the enzyme terminal deoxynucleotidyl transferase.
In the same year Rahman et al. reported the results of their study in which a number of 2’,4’-BNA- and 2’,4’-BNANC-modified siRNAs were designed and synthesized. The thermal stability, nuclease resistance and gene silencing properties against cultured mammalian cells were evaluated and compared with those of natural siRNAs. The 2’,4’-BNA- and 2’,4’-BNANC-modified siRNAs showed very high TM values and were remarkably stable in serum samples. Furthermore, the researchers report that these modified oligo-nucleotides showed promising RNAi properties that were equal to those exhibited by natural siRNAs. The thermally stable siBNAs composed of slightly modified sense and antisense strands suppressed gene expression equal to that of natural siRNA. The modifications at the Argonaut (Ago2) cleavage site of the sense strand (9–11th positions from the 5’-end of the sense strand) produced variable results depending on siRNA composition. However, modification at the 10th position diminished siRNA activity. In moderately modified siRNAs, modification at the 11th position displayed usual RNAi activity, while modification at the 9th position showed variable results depending on the composition of the siRNA.
Yamamoto et al. in 2012 demonstrated that BNA-based antisense therapeutics can be used to successfully inhibit hepatic PCSK9 expression which resulted in a strong reduction of the serum LDL-C levels of mice. These findings support the hypothesis that PCSK9 is a potential therapeutic target for hypercholesterolemia. The researchers state that this is the first time they were able to show that BNA-based antisense oligo-nucleotides (AONs) induced a cholesterol-lowering action in hypercholesterolemic mice. Hypercholesterol-emia is a metabolic condition in which high levels of cholesterol are present in the blood and where elevated levels of lipids and lipoproteins are observed in the blood as well. If untreated higher levels of total cholesterol will increase the risk for cardiovascular disease in particular coronary heart disease.
We expect that BNAs like LNAs will find a widespread use in antisense oligonucleotide technology, where they can be used to stabilize interactions with target RNA and protect them from the attack by cellular nucleases. Furthermore, they can be used in the field of molecular diagnostics and the newly emerging field of siRNAs. Utilizing these modified nucleotides promises to increase double-stranded RNA stability in serum and decrease off-target effects seen with conventional siRNAs. Next, this oligonucleotide technology has the potential to become a new type of therapy to treat a wide variety of diseases, and BNAs will no doubt play a part in future developments of therapeutic and diagnostic oligonucleotides. Comparison of various nucleic acid analogs
Rules for the Design of Probes
Benefits of the BNA technology include: * Ideal for the detection of short RNA and DNA targets * Increases the thermal stability of duplexes * Increases the thermal stability of triplexes * Capable of single nucleotide discrimination * Resistant to exo- and endonucleases resulting in high stability in vivo and in vitro applications * Increased target specificity * Facilitates Tm normalization * Strand invasion properties enable the detection of “hard to access” samples * Compatible with standard enzymatic processes Applications
  References ReferencesBraasch DA, and DR Corey 2001 Locked nucleic acid (LNA): fine-tuning the recognition of DNMA and RNA. Chem Biol 8: 1-7. Braasch DA, Y Liu, and DR Corey 2002 Antisense inhibition of gene expression in cells by oligonucleotides incorporating locked nucleic acids: effect of mRNA target sequence and chimera design. NAR 30: 5160-5167. Grunweller A, E Wyszko, B Bieber,R Jahnel, VA Erdmann, and J Kurreck 2003, Comparison of different antisense strategies in mammalian cells using locked nucleic acids, 2’-O-methyl RNA, phosphorothioates and small interfering RNA. NAR 31: 3185-3193. Hendrix C, H Rosemeyer, B De Bouvere, A Van Aerschot, F Seela, and P Herdewijn 1997 1',5'-Anhydrohexitol oligonucleotides: hybridisation and strand displacement with oligoribonucleotides, interaction with RNase H and HIV reverse transcriptase. Eur J Chem 3: 1513–1520. Yoshiyuki Hari, Satoshi Obika, Mitsuaki Sekiguchi and Takeshi Imanishi; Selective recognition of CG interruption by 20,40-BNA having 1-isoquinolone as a nucleobase in a pyrimidine motif triplex formation. Tetrahedron 59 (2003) 5123–5128. Hyrup B, and PE Nielson 1996 Peptide nucleic acids (PNA): synthesis, properties and potential applications. Bioorg Med Chem 4:5-23. Takeshi Imanishi and Satoshi Obika; BNAs: novel nucleic acid analogs with a bridged sugar moiety. CHEM. COMMUN., 2002, 1653–1659. Johnson MP, LM Haupt, and LR Griffiths 2004 Locked nucleic acids (LNA) single nucleotide polymorphism (SNP) genotype analysis and validation using real-time PCR. NAR 32: e55. Yuuya Kasahara, Shunsuke Kitadume, Kunihiko Morihiro, Masayasu Kuwahara, Hiroaki Ozaki, Hiroaki Sawai, Takeshi Imanishi, Satoshi Obika; Effect of 3’-end capping of aptamer with various 2’,4’-bridged nucleotides: Enzymatic post-modification toward a practical use of polyclonal aptamers. Bioorganic & Medicinal Chemistry Letters 20 (2010) 1626–1629. Harleen Kaur, B. Ravindra Babu, and Souvik Maiti; Perspectives on Chemistry and Therapeutic Applications of Locked Nucleic Acid (LNA). Chem. Rev. 2007, 107, 4672-4697. Tetsuya Kodama,Chieko Matsuo, Hidetsugu Ori, Tetsuya Miyoshi, Satoshi Obika, Kazuyuki Miyashita, Takeshi Imanishi; Design, synthesis, and evaluation of a novel bridged nucleic acid, 2’,5’-BNAON, with S-type sugar conformation fixed by N–O linkage. Tetrahedron 65(2009) 2116–2123. Koshkin AA, SK Singh, P Nielsen, VK Rajwanshi, R Kumar, M Meldgaard, CE Olsen, and J Wengel 1998 LNA (Locked Nucelic Acid): Synthesis of the adenine, cytosine, guanine, 5-methylcytosine, thymine and uracil bicyclonucleoside monomers, oligomerisation, and unprecedented nucleic acid recognition. Tetrahedron 54: 3607-3630. Kurreck J, E Wyszko, C Gillen, and VA Erdmann 2002 Design of antisense oligonucleotides stabilized by locked nucleic acids. NAR 30: 1911-1918. Kvaerno L, and J Wengel 1999 Investigation of restricted backbone conformation as an explanation for the exceptional thermal stabilities of duplexes involving LNA (Locked Nucleic Acid): synthesis and evaluation of abasic LNA. Chem Commun 1999, 7: 657-658. Latorra D, K Campbell, A Wolter, and JM Hurley 2003a Enhanced allele-specific PCR discrimination in SNP genotyping using 3’ locked nucleic acid (LNA) primers. Hum Mut 22: 79-85. Latorra D, K Arar, and JM Hurley 2003b Design considerations and effects of LNA in PCR primers. Mol Cell Probes 17: 253-9. McTigue PM, RJ Peterson, and JD Kahn 2004 Sequence-dependent thermodynamic parameters for locked nucleic acid (LNA)-DNA duplex formation. Biochemistry 43: 5388-5405. Yasunori Mitsuoka, Tetsuya Kodama, Ryo Ohnishi, Yoshiyuki Hari, Takeshi Imanishi and Satoshi Obika; A bridged nucleic acid, 2’,4’-BNACOC: synthesis of fully modified oligonucleotides bearing thymine, 5-methylcytosine, adenine and guanine 2’,4’-BNACOC monomers and RNA-selective nucleic-acid recognition. Nucleic Acids Research, 2009, Vol. 37, No. 4 1225–1238. Note: BNA-COC/DNA and BNA-COC/RNA duplex formation. Kazuyuki Miyashita, S. M. Abdur Rahman, Sayori Seki, Satoshi Obikaab and Takeshi Imanishi; N-Methyl substituted 2’,4’-BNANC: a highly nuclease-resistant nucleic acid analogue with high-affinity RNA selective hybridization. Chem. Commun., 2007, 3765–3767. Nielson PE, and G Haaima 1997 Peptide nucleic acid (PNA). A DNA mimic with a pseudopeptide backbone. Chem Soc Rev 26: 73-78. Nomenclature for polynucleotide chains including for the sugar puckering can be found at: http://www.chem.qmul.ac.uk/iupac/misc/pnuc2.html Satoshi Obika, Daishu Nanbu, Yoshiyuki Hari, Ken.ichiro Morio, Yasuko In, Toshimasa Ishida, and Takeshi Imanishi; Synthesis of 2'-O,4'-C-Methyleneuridine and -cytidine. Novel Bicyclic Nucleosides Having a Fixed C a ,-endo Sugar Puckering. Tetrahedron Letters, Vol. 38, No. 50, pp. 8735-8738, 1997. Obika S, D Nanbu, Y Hari, J-i Andoh, K-i Morio, T Doi, and T Imanishi 1998 Stability and structural features of the duplexes containing nulcoeside analogs with a fixed N-type conformation. 2’-O, 4’-C methylene ribonucleosides. Tetrahedron Lett 39: 5401-5404. Satoshi Obika, Mayumi Onoda, Koji Morita, Jun-ichi Andoh, Makoto Koizumi and Takeshi Imanishi; 3’-Amino-2’,4’-BNA: novel bridged nucleic acids having an N3’->P5’ phosphoramidate linkage. Chem. Commun., 2001, 1992–1993. Note: BNA/DNA; BNA/dsDNA. Satoshi Obika, Yoshiyuki Hari, Mitsuaki Sekiguchi, and Takeshi Imanishi; A 2',4'-Bridged Nucleic Acid Containing 2-Pyridone as a Nucleobase: Efficient Recognition of a C●G Interruption by Triplex Formation with a Pyrimidine Motif. Angew. Chem. Int. Ed. 2001, 40, No. 11, 2079-2081. Satoshi Obika, Mitsuaki Sekiguchi, Roongjang Somjing, and Takeshi Imanishi; Adjustment of the g Dihedral Angle of an Oligonucleotide P3’!N5’ Phosphoramidate Enhances Its Binding Affinity towards Complementary Strands. Angew. Chem. Int. Ed. 2005, 44, 1944 –1947. Satoshi Obika, Masaharu Tomizu, Yoshinori Negoro, Ayako Orita, Osamu Nakagawa, and Takeshi Imanishi; Double-Stranded DNA-Templated Oligonucleotide Digestion Triggered by Triplex Formation. ChemBioChem 2007, 8, 1924 – 1928. Note: Triplex triggered cleavage of oligonucleotides. Satoshi Obika, Hiroyasu Inohara, Yoshiyuki Hari,_ and Takeshi Imanishi; Recognition of T●A interruption by 2’,4’-BNAs bearing heteroaromatic nucleobases through parallel motif triplex formation. Bioorganic & Medicinal Chemistry 16 (2008) 2945–2954. Satoshi Obika, S. M. Abdur Rahman, Bingbing Song, Mayumi Onoda, Makoto Koizumi, Koji Morita, Takeshi Imanishi; Synthesis and properties of 3’-amino-2’,4’-BNA, a bridged nucleic acid with a N3’->P5’ phosphoramidate linkage. Bioorganic & Medicinal Chemistry 16 (2008) 9230–9237. Petersen M, and J Wengel 2003 LNA: a versatile tool for therapeutics and genomics. Trends Biotechnol 21:74-81. Petersen M, CB Nielsen, KE Nielsen, GA Jensen, K Bondensgaard, SJ Singh, VK Rajwanshi, AA Koshkin, BM Dahl, J Wengel, and JP Jacobsen 2000 The conformations of locked nucleic acids (LNA). J Mol Recognition 13: 44-53. Petersen M, JJ Sorensen, and JT Nielsen 2003 Structural basis for LNA (locked nucleic acid) triplex formation. Presented at the 5th International Congress on Molecular Structural Biology. S. M. Abdur Rahman, Sayori Seki, Satoshi Obika, Sunao Haitani, Kazuyuki Miyashita, and Takeshi Imanishi; Highly Stable Pyrimidine-Motif Triplex Formation at Physiological pH Values by a Bridged Nucleic Acid Analogue. Angew. Chem. Int. Ed. 2007, 46, 4306 –4309. S. M. Abdur Rahman, Sayori Seki, Satoshi Obika, Haruhisa Yoshikawa, Kazuyuki Miyashita, and Takeshi Imanishi; Design, Synthesis, and Properties of 2’,4’-BNANC: A Bridged Nucleic Acid Analogue.J. AM. CHEM. SOC. 2008, 130, 4886-4896. S. M. Abdur Rahman, Hiroyuki Sato, Naoto Tsuda, Sunao Haitani, Keisuke Narukawa, Takeshi Imanishi, Satoshi Obika; RNA interference with 2’,4’-bridged nucleic acid analogues. Bioorganic & Medicinal Chemistry 18 (2010) 3474–3480. Schulz RG, and SM Gryaznov 1996 Oligo-2'-fluoro-2'-deoxynucleotide N3'-->P5' phosphoramidates: synthesis and properties. NAR 24: 2966-73. Simeonov A, and TT Nikiforov 2002 Single nucleotide polymorphism genotyping using short, fluorescently labeled locked nucleic acid (LNA) probes and fluorescence polarization detection. NAR 30: e91. Singh SK, P Nielsen, AA Koshkin, and J Wengel 1998 LNA (locked nucleic acids): synthesis and high-affinity nucleic acid recognition. Chem Commun 4: 455-456. Tolstrup N, PS Nielsen, JG Kolberg, AM Frankel, H Vissing, and S Kauppinen 2003 OilgoDesign: optimal design of LNA (locked nucleic acid) oligonucleotide capture probes for gene expression profiling. NAR 31: 3758-3762. Torigoe H, Y Hari, M Sekiguchi, S Obika, and T Imanishi 2001; 2’-O, 4’-C-methylene bridged nucleic acid modification promotes pyrimidine motif triplex DNA formation at physiologic pH. J Biol Chem 276: 2354-2360. Note: TFO for therapeutics. Hidetaka Torigoe, Osamu Nakagawa, Takeshi Imanishi, Satoshi Obika, Kiyomi Sasaki; Chemical modification of triplex-forming oligonucleotide to promote pyrimidine motif triplex formation at physiological pH. Biochimie 94 (2012) 1032-1040. Note: pyrimidine motif triplex formation by 3’-amino-2’-O,4’-BNA. Ugozzoli LA, D Latorra, R Pucket, K Arar, and K Hamby 2004 Real-time genotyping with oligonucleotide probes containing locked nucleic acids. Anal Biochem 324: 143-152. Van Aerschot A, I Verheggen, C Hendrix,and P Herdewijn 1995 1,5-Anhydrohexitol nucleic acids, a new promising antisense construct. Angew Chem Int Ed 34: 1338–1339. Tsuyoshi Yamamoto, Mariko Harada-Shiba, Moeka Nakatani, Shunsuke Wada, Hidenori Yasuhara, Keisuke Narukawa, Kiyomi Sasaki, Masa-Aki Shibata, Hidetaka Torigoe, Tetsuji Yamaoka, Takeshi Imanishi and Satoshi Obika; Cholesterol-lowering Action of BNA-based Antisense Oligonucleotides Targeting PCSK9 in Atherogenic Diet-induced Hypercholesterolemic Mice Molecular Therapy–Nucleic Acids (2012) 1, e22; oi:10.1038/mtna.2012.16. |
 Structures of Watson-Crick-type base pairs, G•C and A•T (left), and Hoogsteen type base triads, C+•G•C and T•A•T (right).
Structures of Watson-Crick-type base pairs, G•C and A•T (left), and Hoogsteen type base triads, C+•G•C and T•A•T (right).

 Monomer structures of BNAs. The structures of selected nucleic acid analogs with a bridged sugar moiety are depicted.
Monomer structures of BNAs. The structures of selected nucleic acid analogs with a bridged sugar moiety are depicted. The chemical structures and the energy minimized molecular models for the 2’, 4’-BNA-NC[NH] and 2’, 4’-BNA-NC[NCH3] monomers are illustrated here.
The chemical structures and the energy minimized molecular models for the 2’, 4’-BNA-NC[NH] and 2’, 4’-BNA-NC[NCH3] monomers are illustrated here.↧
↧
Citrullinated Peptide Synthesis
Citrullinated Peptide Synthesis
The amino acid citrulline is not coded for by DNA directly however several proteins are known to contain citrulline as a result of a posttranslational modification. These citrulline residues are generated by a family of enzymes called peptidylarginine deiminases (PADs), which convert arginine into citrulline in a process called citrullination or deimination. It has became clear in recent years that histones, fibronectin, myelin basic protein (MBP) as well as other cellular proteins can be modified by post-translational changes during epigenetic regulation in the cell. Modifications include acetylation, methylation, phosphorylation, ubiquitination and citrullination among others. Histone modifications induce changes to the struc&not;ture of chromatin and thereby affect the accessibility of the DNA strand to transcriptional enzymes, resulting in activation or repression of genes associated with modified histones. So far citrulline modifications have been connected with the autoimmune disorders multiple sclerosis and rheumatoid arthritis. Biosynthesis offers citrulline incorporation into any peptide sequence to help researchers study the effect of these modifications in vivo or in vitro.
Deimination reaction of arginyl residues within peptide bonds Deimination of arginine results in neutral citrulline with the release of ammonia and the loss of one positive charge for each arginyl residue deiminated. The process is catalyzed by a calcium dependent peptidylarginine deiminase.
Contact us for more information on Peptide Citrullin modification
↧
Mycobacterium tuberculosis peptide libraries
Mycobacterium tuberculosis peptide librariesDesign of Mtb peptide libraries for targeted research Tuberculosis, an ancient disease, has become an escalating global health problem in recent years. It is estimated that one-third of the world’s population presently is infected with Mycobacterium tuberculosis (Mtb) and that the bacteria causes 1.7 million deaths annually. On November 20th, 2012 the news reported that students in Grand Forks, ND, were diagnosed with tuberculosis and health officials reported three new cases of tuberculosis (TB) in Grand Forks, including a student at Valley Middle School. This brought the total number of active cases to 13. An article in the Wall Street Journal on Friday November 23, 2012 reported how the fight against TB made the bacteria stronger. Basically, the use of the standard treatment to cure TB in India and other countries with a high infection rate by giving patients a cocktail of antibiotic drugs at a cost of $9 per month without prior testing for drug-resistant strains, allowed the bacteria to evolve more resistant strains. Additional to the cost for testing in a certified lab to help guide a successful treatment, the cost to treat resistant TB strains amounts to $2,000 per month. Chances are that until an integrated approach that uses testing for drug-resistance together with a targeted treatment is established, the infection rate of TB will continue to increase worldwide.
1. Non over lapping peptide libraries:
The library for this selected protein consists of 16 peptides. The selected peptides can be modified either on the N- or C-terminal end with tyrosine to allow for the use of I125 as the radiolabel, cysteine, to allow for conjugation to another compound or to beads, biotin, to allow for binding to avidin or streptavidin coated beads, or any other desired label such as a fluorophore or stable isotope tag, to allow for the coding of the peptides. The use for the downstream analysis tools will determine what type of label needs to be selected.
Many more permutations of peptide library designs are possible. The resulting libraries can be used to screen highly active compounds such as antigenic peptides, receptor ligands, antimicrobial compounds, protein binding interfaces, and enzyme inhibitors among others. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
immune cells known as macrophages, can become further stratified by the formation of a fibrous extra layer of extracellular matrix material that is laid down outside the macrophage layer. Lymphocytes appear to be restricted primarily to this peripheral area. Many of the granulomas persist in this balanced state, but progression toward disease is characterized by the loss of vascularization, increased necrosis, and the accumulation of caseum in the granuloma center. Ultimately, infectious bacilli are released into the airways when the granuloma cavitates and collapses into the lungs. (Taken from Russel at al. 2010). According to the paper the research field is lacking some of the most basic tools to evaluate or assessing the beneficial effect of new drugs. Furthermore, no clear biomarkers to assess the disease status are presently available.
Chun et al. in 2001 report the use of N-formylated Mtb peptides to test their ability to bind M3 using an immunofluorescence-based peptide-binding assay. M3 is an MHC class Ib molecule that preferentially presents N-formulated peptides to CD8+ T cells. Bacteria initiate protein synthesis with N-formylated methionine which makes M3 especially suitable for presenting this types of peptide epitopes. Therefore, the research group scanned the full sequence of the Mtb genome for N-terminal peptides that shared common features with other M3-binding peptides using bioinformatic tools. Synthetic peptides corresponding to the selected sequences were tested for their ability to bind M3. Furthermore, the researchers report that four of these peptides were able to elicit cytotoxic T lymphocytes (CTLs) from mice immunized with peptide-coated splenocytes. They concluded that their data suggest that M3-restricted T cells may participate in the immune response to Mtb.
Taehoon Chun, Natalya V. Serbina, Dawn Nolt, Bin Wang, Nancy M. Chiu, JoAnne L. Flynn, and Chyung-Ru Wang; Induction of M3-restricted Cytotoxic T Lymphocyte Responses by N-formylated Peptides Derived from Mycobacterium tuberculosis. J. Exp. Med. Volume 193, Number 10, May 21, 2001 1213–1220. Christopher Dye and Brian G. Williams; The Population Dynamics and Control of Tuberculosis. Science 328, 856 (2010). Simone A. Joosten, Krista E. van Meijgaarden, Pascale C. van Weeren, Fatima Kazi, Annemieke Geluk, Nigel D. L. Savage; Mycobacterium tuberculosis Peptides Presented by HLA-E Molecules Are Targets for Human CD8+ T-Cells with Cytotoxic as well as Regulatory Activity. PLoS Pathogens | www.plospathogens.org 1 February 2010 | Volume 6 | Issue 2 | e1000782. Riva Kovjazina, Ilan Volovitzc, Yair Daona, Tal ViderShalitb, Roy Azranb, Lea Tsabanb, Lior Carmona, Yoram Louzoun; Signal peptides and transmembrane regions are broadly immunogenic and have high CD8+ T cell epitope densities: Implications for vaccine development. Molecular Immunology 48 (2011) 1009–1018. Frieder M, Lewinsohn DM. T-cell epitope mapping in Mycobacterium tuberculosis using pepmixes created by micro-scale SPOT- synthesis. Methods Mol Biol. 2009;524:369-82. Marisol Ocampo, Daniel Aristiza´bal-Ramı´rez, Diana M.Rodrı´guez, Marina Mun˜oz, Hernando Curtidor, Magnolia Vanegas, Manuel A.Patarroyo, and Manuel E.Patarroyo; The role of Mycobacterium tuberculosis Rv3166c protein-derived high-activity binding peptides in inhibiting invasion of human cell lines. Protein Engineering, Design & Selection vol. 25 no. 5 pp. 235–242, 2012. David G. Russell, Clifton E. Barry 3rd, JoAnne L. Flynn; Tuberculosis: What We Don't Know Can, and Does, Hurt Us. Science 328, 852 (2010). Winslow GM, Cooper A, Reiley W, Chatterjee M, Woodland DL; Early T-cell responses in tuberculosis immunity. Immunol Rev. 2008 Oct; 225:284-99. Wirth T, Hildebrand F, Allix-Béguec C, Wölbeling F, Kubica T, et al. (2008) Origin, Spread and Demography of the Mycobacterium tuberculosis Complex. PLoS Pathog 4(9): e1000160. doi:10.1371/journal.ppat.1000160
|


↧
Brief history of Bridged Nucleic Acids (BNAs)
A brief history of Bridged Nucleic Acids (BNAs)A quest for better oligonucleotide mimics. The quest for oligonucleotide mimics with improved characteristics and stabilities useful for molecular diagnostics and therapeutics that also show minimal side effects has led to the design and synthesis of novel bridged nucleic acid monomers and oligonucleotides. These synthetic oligonucleotide mimics may be used as tools for gene validation, as antisense (targeting mRNA) and antigene (targeting DNA) agents, for selective regulation of gene expression and as a potentially new class of drugs for the treatment of diseases such as cancer, inflammation, viral diseases and other pathological diseases. The 3D structures for A-RNA and B-DNA were used as a template for the design of the BNA monomers. The goal for the design is to find derivatives that posses high binding affinities with complementary RNA and/or DNA strands.
| |||||
  | |||||
Synthetic oligonucleotides are now important, established tools for life scientists and have many applications in molecular biology and genetic diagnostics, and are poised to become important tools in the emerging field of molecular medicine as well. While unmodified oligodeoxynucleotides can form DNA:DNA and DNA:RNA duplexes they are sometimes unstable and labile to nucleases. Therefore a variety of nucleic acid analogs have been developed to enhance high-affinity recognition of DNA and RNA targets, enhancing duplex stability and assist with cellular uptake.
Bridged nucleic acids (BNAs) are molecules that contain a five-membered or six-membered bridged structure with a “fixed” C3’-endo sugar puckering (Saenger 1984). The bridge is synthetically incorporated at the 2’, 4’-position of the ribose to afford a 2’, 4’-BNA monomer. The monomers can be incorporated into oligonucleotide polymeric structures using standard phosphoamidite chemistry. BNAs are structurally rigid oligo-nucleotides with increased binding affinities and stability. The incorporation of BNAs into oligonucleotides allows the production of modified synthetic oligonucleotides with:
BNAs can be synthesized using standard phosphoramidite chemistry. The first synthesis of bridged 2’-O, 4’-C-methyleneuridine and –cytidine monomers were described by Obika et al. in 1997 (in Imanishi’s group). The same group showed in 1998 that these monomers allowed the formation of stable oligonucleotide duplexes in both DNA and RNA based synthetic 12 meric oligonucleotides. Chemical structures for nucleosides and a bridged nucleoside are shown below. | |||||
 | |||||
Koshkin et al. in 1998 demonstrated that these monomers can be used to synthesize oligonucleotides that can form stable complexes with DNA and RNA oligonucleotides. Furthermore, the group gave these monomers a new name and called them “Locked Nucleic Acids” (LNAs). The synthesis of these bridged nucleic acids could be achieved by standard phosphoramidite chemistry. Jesper Wengel in 1999 describes the synthesis of 3’-C- and 4’-C-branched oligonucleotides and the development of locked nucleic acids as well as their use as DNA/RNA mimics. Christensen et al. in 2001 used stopped-flow kinetic measurements to study the thermodynamics of LNA oligonucleotide based complexes. Obika at al. in 2001 report that 2'-O, 4'-C-methylene bridged nucleic acids (2',4'-BNAs = LNA) have unprecedented binding affinities towards their complementary RNA. The researchers showed that 2',4'-BNA oligonucleotides can be used as antisense molecules and demonstrated their potent inhibitory effect on gene expression of Intercellular Adhesion Molecule-1 (ICAM-1) in living cells. Furthermore, the contribution of RNase H to this antisense effect and adequate stability of 2',4'-BNA oligonucleotides to enzymatic degradation were also demonstrated. These results showed that BNAs can be used to find natural RNA sequences and target them for destruction. Torigoe et al. also in 2001 report that 2'-O, 4'-C-methylene bridged nucleic acids (LNAs) can be used to synthesize modified oligonucleotides that can form triplexes with DNA at physiological pH. LNAs are the best studied and characterized bridged nucleic acids so far.
Also in 2001 Obika et al. introduced a 3’-amino-2’,4’-BNA monomer and a 2’,4’-BNA that contained a 2-pyridone group as the base that showed duplex and triplex forming abilities when used in oligonucleotides. | |||||
 | |||||
Another bridged nucleic acid monomer was synthesized and introduced in 2001 by Morita et al. called 2’-O, 4’-C-ethylene-bridged nucleic acid (ENA). The 2'-O,4'-C-ethylene linkage of these nucleosides restricts the sugar puckering to the N-conformation. The ethylene-bridged nucleic acids showed a high binding affinity for the complementary RNA strand (ΔTm = +5°C per modification) and were approximately 400 and 80 times more nuclease-resistant than natural DNA and BNA/LNA, respectively. These results indicate that ENA have better antisense activities than BNA/LNA.
Hari et al. in 2003 developed a novel nucleoside analogue that allowed for the effective recognition of CG interruption in a homopurine–homopyrimidine tract of double-stranded DNA (dsDNA). The scientists succeeded in the synthesis of a triplex-forming oligonucleotide (TFO) containing the novel 2’,4’-BNA (QB) bearing 1-isoquinolone as a nucleobase. The triplex-forming ability and sequence-selectivity of the TFO (TFO-QB) were examined. Melting temperature (Tm) measurements found that the TFO-QB formed a stable triplex DNA in a highly sequence-selective manner under near physiological conditions.
Tolstop et al. in 2003 published a paper that described a software tool called “OligoDesign” that allowed for the ‘in-silico” design of LNA based oligonucleotides. The software provides optimal design of LNA (locked nucleic acid) substituted oligonucleotides for functional genomics applications. The OligoDesign software features recognition and filtering of the target sequence by genome-wide BLAST analysis in order to minimize cross-hybridization with non-target sequences. Routines for prediction of melting temperature, self-annealing and secondary structure for LNA substituted oligonucleotides, as well as secondary structure prediction of the target nucleotide sequence are included. Individual scores for all these properties are calculated for each possible LNA oligonucleotide in the query gene and the OligoDesign program ranks the LNA capture probes according to a combined fuzzy logic score and finally returns the top scoring probes to the user in the output. The OligoDesign program is freely accessible at http://lnatools.com/
The bioinformatics tools was designed to optimize the design of modified oligonucleotides used for the following applications:
Antisense oligonucleotides that contain LNAs show improved silencing potency but cause significant hepatoxicity in animals. This was noticed in 2006 by Swayze at el. when designing antisense oligonucleotides for the silencing of TRADD and ApoB genes in cell cultures. These results indicated that LNAs may need to be used with caution for antisense purposes. These characteristics led to design newer generations of BNAs. Miyashita et al. in 2007 (in Imanishi’s group) report the design and synthesis of a new type of BNA, a N-methyl substituted 2’,4’-BNANC. This is a highly nuclease-resistant nucleic acid analogue with high-affinity RNA selective hybridization. The monomer was designed to fine tune the BNA structure. | |||||
| |||||
The research group synthesized a novel bridged nucleic acid 2’,4’-BNANC[N–Me] and showed that it has high-affinity hybridization similar to that of 2’,4’-BNA (LNA) against an RNA complement. Furthermore, the scientists report that, the nucleic acid analogue displayed RNA selectivity superior to that of 2’,4’-BNA (LNA) and other structural analogues of 2’,4’-BNA (LNA). Nuclease resistance of this nucleic acid analogue is abundantly higher than that of 2’,4’-BNA (LNA) and also slightly higher than that of a phosphorthioate. The hydrophobic methyl substituent on the backbone might present an additional advantage resulting in cellular uptake of the oligonucleotides. All of these reported characteristics of the BNA are essential for antisense applications. In the same year Rahman et al. report that 2’,4’-BNANC form highly stable pyrimidine-motif DNA triplexes at physiological pH. These triplexes are involved in the regulation of gene expression, site-specific cleavage of DNA, gene mapping and isolation, maintenance of folded chromosome confromations, and gene-targeted mutagenesis. In a pyrimidine-motif triplex DNA the triplex forming oligonucleotide binds with the homopurine tract of the target duplex DNA in a sequence specific manner through Hoogsteen hydrogen bonds to form T●A:T and C+●G:C triads. In the same year Obika et al report that 5’-amino-BNAs can be used to digest oligonucleotides triggered by triplex formation.
In 2008 Imanishi’s group (Rahman et al. 2008) introduced three new bridged nucleic acid analogues called 2’,4’-BNANC[NH], 2’,4’-BNANC[NMe], and 2’,4’-BNANC[NBn]. Structures of these analogs are shown below. The new analogs were designed by taking the length of the bridged moiety into account. A six-membered bridged structure with a unique structural feature (N-O bond) in the sugar moiety was designed to have a nitrogen atom. This atom can act as a conjugation site and improve the formation of duplexes and triplexes by lowering the repulsion between the negatively charged backbone phosphates. Furthermore, the nitrogen atom on the bridge can be functionalized by hydrophobic and hydrophilic groups, by adding groups that introduce steric bulk or any desired functional moiety. These modifications allow to control affinity towards complementary strands, regulate resistance against nuclease degradation and the synthesis of functional molecules designed for specific applications in genomics. The properties of these analogs were investigated and compared to those of previous 2’,4’-BNA (LNA) modified oligonucleotides. | |||||
| |||||
Compared to 2’,4’-BNA (LNA)-modified oligonucleotides, 2’,4’-BNANC congeners were found to possess:
The researchers state that “2’,4’-BNANC-modified oligonucleotides with these excellent profiles show great promise for applications in antisense and antigene technologies.” More recently Yamamoto et al. in 2012 demonstrated successfully that BNA-based antisense therapeutics inhibited hepatic PCSK9 expression, resulting in a strong reduction of the serum LDL-C levels of mice. The findings support the hypothesis that PCSK9 is a potential therapeutic target for hypercholesterolemia. This appears to be the first time that researchers were able to show that BNA-based antisense oligonucleotides (AONs) induced cholesterol-lowering action in hypercholesterolemic mice. A moderate increase of aspartate aminotransferase, ALT, and blood urea nitrogen levels was observed whereas the histopathological analysis revealed no severe hepatic toxicities. The same group, also in 2012, report that the 2’,4’-BNANC[NMe] analog when used in antisense oligonucleotides showed significantly stronger inhibitory activities which is more pronounced in shorter (13- to 16mer) oligonucleotides. Their data led the researchers to conclude that the 2’,4’-BNANC[NMe] analog may be a better alternative to conventional LNAs. Action mechanism of antisense oligonucleotides The proposed action mechanism for antisense oligonucleotides may involve translation arrest, mRNA degradation mediated by RNase H and splicing arrest. This is illustrated in the following figure.
| |||||
| |||||
References Ulla CHRISTENSEN, Nana JACOBSEN., Vivek K. RAJWANSHI, Jesper WENGEL and Troels KOCH. Stopped-flow kinetics of locked nucleic acid (LNA)–oligonucleotide duplex formation : studies of LNA–DNA and DNA–DNA interactions Biochem. J. (2001) 354, 481-484. Yoshiyuki Hari, Satoshi Obika, Mitsuaki Sekiguchi and Takeshi Imanishi; Selective recognition of CG interruption by 2’,4’-BNA having 1-isoquinolone as a nucleobase in a pyrimidine motif triplex formationqTetrahedron 59 (2003) 5123–5128. Makoto KOIZUMI; 2’-O,4’-C-Ethylene-Bridged Nucleic Acids (ENATM) as Next-Generation Antisense and Antigene Agents. Biol. Pharm. Bull. 27(4) 453-456 (2004). Koizumi M.; ENA oligonucleotides as therapeutics. Curr Opin Mol Ther. 2006 Apr;8(2):144-9. Koshkin AA, SK Singh, P Nielsen, VK Rajwanshi, R Kumar, M Meldgaard, CE Olsen, and J Wengel 1998 LNA (Locked Nucelic Acid): Synthesis of the adenine, cytosine, guanine, 5-methylcytosine, thymine and uracil bicyclonucleoside monomers, oligomerisation, and unprecedented nucleic acid recognition. Tetrahedron 54: 3607-3630. Koshkin, A.A., Nielsen, P., Meldgaard, M., Rajwanshi, V. K., Singh S. K. and Wengel, J. (1998). LNA (Locked Nucleic Acid): An RNA Mimic Forming Exceedingly Stable LNA:LNA Duplexes. J. Am. Chem. Soc. 120, 13252 – 13253. Kazuyuki Miyashita, S. M. Abdur Rahman, Sayori Seki, Satoshi Obikaab and Takeshi Imanishi; N-Methyl substituted 2’,4’-BNANC: a highly nuclease-resistant nucleic acid analogue with high-affinity RNA selective hybridization. Chem. Commun., 2007, 3765–3767. Koji Morita, Chikako Hasegawa, Masakatsu Kaneko, Shinya Tsutsum P, Junko Sone, Tomio Ishikawa, Takeshi Imanishi and Makoto Koizumi; 2'-O,4'-C-Ethylene-bridged nucleic acids (ENA) with nuclease resistance and high affinity for RNA. 2001. Nucleic Acids Research Supplement No. 1 241-242. Nomenclature for polynucleotide chains including for the sugar puckering can be found at: http://www.chem.qmul.ac.uk/iupac/misc/pnuc2.html Satoshi Obika, Daishu Nanbu, Yoshiyuki Hari, Ken.ichiro Morio, Yasuko In, Toshimasa Ishida, and Takeshi Imanishi; Synthesis of 2'-O,4'-C-Methyleneuridine and -cytidine. Novel Bicyclic Nucleosides Having a Fixed C3 ,-endo Sugar Puckering. Tetrahedron Letters, Vol. 38, No. 50, pp. 8735-8738, 1997. Obika S, D Nanbu, Y Hari, J-i Andoh, K-i Morio, T Doi, and T Imanishi 1998. Stability and structural features of the duplexes containing nulcoeside analogs with a fixed N-type conformation. 2’-O, 4’-C methylene ribonucleosides. Tetrahedron Lett 39: 5401-5404. Satoshi Obika,Mayumi Onoda,Koji Morita,Jun-ichi Andoh,Makoto Koizumiand Takeshi Imanishi; 3’-Amino-2’,4’-BNA: novel bridged nucleic acids having an N3’->P5’ phosphoramidate linkage. Chem. Commun., 2001, 1992–1993. Note: BNA/DNA; BNA/dsDNA. Satoshi Obika, Yoshiyuki Hari, Mitsuaki Sekiguchi, and Takeshi Imanishi; A 2',4'-Bridged Nucleic Acid Containing 2-Pyridone as a Nucleobase: Efficient Recognition of a C●G Interruption by Triplex Formation with a Pyrimidine Motif. Angew. Chem. Int. Ed. 2001, 40, No. 11, 2079-2081. Obika S, Hemamayi R, Masuda T, Sugimoto T, Nakagawa S, Mayumi T, Imanishi T.; Inhibition of ICAM-1 gene expression by antisense 2',4'-BNA oligonucleotides. Nucleic Acids Res Suppl. 2001;(1):145-6. Satoshi Obika, Masaharu Tomizu, Yoshinori Negoro, Ayako Orita, Osamu Nakagawa, and Takeshi Imanishi; Double-Stranded DNA-Templated Oligonucleotide Digestion Triggered by Triplex Formation. ChemBioChem 2007, 8, 1924 – 1928. Note: Triplex triggered cleavage of oligonucleotides. S. M. Abdur Rahman, Sayori Seki, Satoshi Obika, Sunao Haitani, Kazuyuki Miyashita, and Takeshi Imanishi; Highly Stable Pyrimidine-Motif Triplex Formation at Physiological pH Values by a Bridged Nucleic Acid Analogue. Angew. Chem. Int. Ed. 2007, 46, 4306 –4309. Saenger, W.; Principles of Nucleic Acid Structure, Springer-Verlag, new York, 1984. Eric E. Swayze,* Andrew M. Siwkowski, Edward V. Wancewicz, Michael T. Migawa, Tadeusz K. Wyrzykiewicz, Gene Hung, Brett P. Monia, and C. Frank Bennett; Antisense oligonucleotides containing locked nucleic acid improve potency but cause significant hepatotoxicity in animals Nucleic Acids Res. 2007 January; 35(2): 687–700. Torigoe H, Y Hari, M Sekiguchi, S Obika, and T Imanishi 2001; 2’-O, 4’-C-methylene bridged nucleic acid modification promotes pyrimidine motif triplex DNA formation at physiologic pH. J Biol Chem 276: 2354-2360. Note: TFO for therapeutics. Tsuyoshi Yamamoto, Mariko Harada-Shiba, Moeka Nakatani, Shunsuke Wada, Hidenori Yasuhara, Keisuke Narukawa, Kiyomi Sasaki, Masa-Aki Shibata, Hidetaka Torigoe, Tetsuji Yamaoka, Takeshi Imanishi and Satoshi Obika; Cholesterol-lowering Action of BNA-based Antisense Oligonucleotides Targeting PCSK9 in Atherogenic Diet-induced Hypercholesterolemic Mice Molecular Therapy–Nucleic Acids (2012) 1, e22; oi:10.1038/mtna.2012.16. Tsuyoshi Yamamoto, Hidenori Yasuhara, FumitoWada, Mariko Harada-Shiba, Takeshi Imanishi, and Satoshi Obika; Superior Silencing by 2’,4’-BNANC-Based Short Antisense Oligonucleotides Compared to 2’,4’-BNA/LNA-Based Apolipoprotein B Antisense Inhibitors. Hindawi Publishing Corporation, Journal of Nucleic Acids Volume 2012, Article ID 707323, 7 pages doi:10.1155/2012/707323. Yong You, Bernardo G. Moreira, Mark A. Behlke, and Richard Owczarzy; Design of LNA probes that improve mismatch discrimination. Nucleic Acids Research, 2006, Vol. 34, No. 8 e60. | |||||





↧
DNA-mediated site-specific deposition of gold nanoparticles on silicon wafers
We report a method for the site-specific deposition of gold nanoparticles, as programmed by DNA sequences immobilized on the surface of silicon oxide-coated silicon wafers. After optimization of surface chemistries, selectivities of between 8 : 1 and 118 : 1 were achieved for the DNA-based sorting of populations of gold nanoparticle of 15 nm and 60 nm diameter from a common suspension via oligonucleotide duplex formation.
↧
↧
Bridged Nucleic Acid (BNA3) Design Guidelines
Design of BNANC [NMe] modified oligonucleotidesThe placing of BNA NC [NMe] monomers in oligonucleotides, both, for DNA and RNA, is shown based on examples from published experimental data!
N = position of BNA NC [NMe] monomer.
1. Gene silencing by targeting mRNA usingBNANC[NMe]’s ! Design of BNANC oligonucleotide: 5’-NNNnnnnnnnnNNN-3’, where N depicts the position of a BNAnc monomer, and n depicts the position of a natural DNA. The use of BNANC’s increased nuclease resistance and improved overall properties for gene silencing by targeting mRNA. Buffer used: 100 mM NaCl, 10 mM sodium phosphate pH 7.2 for thermal melting studies. In vitro transfection was done with Lipofetamine 2000. 2′,4′-BNANC–based AONs are promising therapeutic agents for antisense therapy!! BNANC’s for gene silencing by mRNA targeting! Yamamoto et al. 2012 showed that 2’,4’-BNANC-based AONs targeting apoB mRNA have higher binding affinities to the target RNA than do 2’,4’-BNA/LNA-based AONs. Additionally, in vitro transfection studies revealed the superior silencing effect of short 2’,4’-BNANC-based AONs (<20-ntlong), indicating that 2’,4’-BNANC may have advantageous properties as short antisense drugs. The BNANCs corresponding to the LNAs showed stronger inhibitory activities. Shorter AONs (13- to 14mers) showed better inhibitory activities. The 2’,4’-BNANC-14mer worked the best. 2. Gene silencing by targeting mRNA! Design of BNANC oligonucleotide: 5′-NNnnnNNNnNnnnnnNnNNn-3′ Yamamoto et al. 2012: In vitro and in-vivo study. BNANC[NMe]’s with cholesterol lowering action targeting PCSK9 and with increased nuclease resistance and improved properties for gene silencing by targeting mRNA. Yamamoto and colleges achieved a dose-dependent decrease in serum LDL-C levels by using a 2′,4′-BNANC–based AON (P901SNC). Serum HDL-C levels and the levels of liver and kidney toxicity indicators were not elevated. Histopathological analysis revealed no severe hepatic toxicities. They also showed that a 2′,4′-BNANC–based AON (P901SNC) has greater potential to inhibit PCSK9 and to reduce serum cholesterol levels with no toxicity than a conventional 2′,4′-BNA–based AON. The high-potency and low-toxicity characteristics of a 2′,4′-BNANC–based AON were previously reported to effectively inhibit PTEN mRNA without elevation of the serum ALT level, whereas elevated serum ALT was observed in the 2′,4′-BNA counterpart-treated arm. Thus, it was concluded that 2′,4′-BNANC–based AONs can be a promising therapeutic agent for antisense therapy. 3. Interference RNA: => siRNA: BNANC’s for siRNA - RNAi => siRNA Design of BNANC oligonucleotides that worked best: 5’-NNNnNnnnnnnnnnnnnnnnn-3’, 5’-nnNnnnNnnnNnnnNnnnNnn-3’ 5’-nnnnnnnnnnNnNNnnnnnnn-3’, 5’-nnNnnNnnnnNnnnnNnnnnn-3’ Rahman and colleges in 2010 found that 2’,4’-BNA- and 2’,4’-BNANC-modified siRNAs are equally compatible with the RNAi machinery similar to that observed for natural siRNA. To improve siRNA biostability, a number of bridged nucleotide moieties can be incorporated in the sense strand without loss of the usual gene silencing property. Thermally stable functional siRNAs can also be obtained by slightly modifying the middle of the sense and antisense strands together. Unlike the 3’-overhang modification, this modification increased Tm satisfactorily and contains an antisense strand with BNA residues which might be more efficacious in gene silencing. Modification at the Ago2 cleavage site (9–11th positions) produced variable results based on siRNA composition and sequence; usually the modification at the 10th position of the sense strand is more sensitive. Modification at the 11th position of the cleavage site is safer than that of the 10th or 9th position. For the first time, this study as a whole shows the utility and capability of 2’,4’-BNANC, a highly stable and efficient nucleic acid derivative in RNAi technology, and also gives some new ideas about designing biostable, functional siRNAs consisting both of 2’,4’-BNA and 2’,4’-BNANC residues. 4. Antisense oligonucleotide targeting PTEN mRNA Design of BNANC oligonucleotide: 5’-NNnnnnnnnnnnNN-3’ 14mer 43 d(CUTAGCACTGGCCU)30 20,40-BNANC[NMe] PTEN Prakash et al in 2010. 5. Aptamer Capping at the 3’-end A thrombin aptamer was capped with BNAs at the 3’-end. This increased nuclease resistance and the stability of the aptamer. Kasahara et al. 2010. 6. Synthesis of Novel 2’,4’-BNANC [NMe] nucleic acid analog. Duplex formation: 5-d(GCGTTTTTTGCT) Target ssRNA 5’-r(AGCAAAAAACGT)-3’, Tm = 63 Target ssdNA 5’-r(AGCAAAAAACGT)-3’, Tm = 51 Triplex formation: 5-d(TTTTTCTTTCTCT) 2’,4’-BNANC [NH] and LNA are better. Target: dsDNA 5’-GCTAAAAAGAAAGAGAGATCG)-3’ 3’-CGATTTTTCTTTCTCTCTCTAGC)-5’ Rahman et al. 2008.
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||


↧
Allele Specific BNA Primer Design
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||



↧
Peptide libraries for heterogeneous nuclear ribonucleoproteins or hnRNPs
Heterogeneous nuclear ribonucleoproteins (hnRNPs) isoformsHeterogeneous nuclear ribonucleoproteins (hnRNPs) comprise a family of RNA-binding proteins. The nature of hnRNPs is very complex and diverse. This multifunctional protein family is involved not only in processing heterogeneous nuclear RNAs (hnRNAs) into mature mRNAs, but also acting as trans-factors in regulating gene expression. Heterogeneous nuclear ribonucleoprotein E1 (hnRNP E1), a subgroup of hnRNPs, is a KH-triple repeat containing the RNA-binding protein. It is encoded by an intronless gene arising from hnRNP E2 through a retrotransposition event. hnRNP E1 is ubiquitously expressed and functions in regulating major steps of gene expression, including pre-mRNA processing, mRNA stability, and translation. This protein family exhibit wide-ranging functions in the nucleus and cytoplasm and interaction with multiple other proteins and appears to be involved in post-transcriptional regulation events or pathways in eukaryotic organisms. The A/B subfamily of ubiquitously expressed hnRNPsThis gene belongs to the A/B subfamily of ubiquitously expressed heterogeneous nuclear ribonucleo-proteins (hnRNPs). The hnRNPs are RNA binding proteins and complex with heterogeneous nuclear RNA (hnRNA). These proteins are associated with pre-mRNAs in the nucleus and appear to influence pre-mRNA processing and other aspects of mRNA metabolism and transport. While all of the hnRNPs are present in the nucleus, some seem to shuttle between the nucleus and the cytoplasm. The hnRNP proteins have distinct nucleic acid binding properties. The protein encoded by this gene has two repeats of quasi-RRM domains that bind to RNAs. It is one of the most abundant core proteins of hnRNP complexes and it is localized to the nucleoplasm. This protein, along with other hnRNP proteins, is exported from the nucleus, probably bound to mRNA, and is immediately re-imported. Its M9 domain acts as both a nuclear localization and nuclear export signal. The encoded protein is involved in the packaging of pre-mRNA into hnRNP particles, transport of poly A+ mRNA from the nucleus to the cytoplasm, and may modulate splice site selection. It is also thought to have a primary role in the formation of specific myometrial protein species in childbirth. [The myometrium is the middle layer of the uterine wall, mainly consisting of uterine smooth muscle cells.] Multiple alternatively spliced transcript variants have been found for this gene but only two transcripts are fully described. These variants have multiple alternative transcription initiation sites and multiple polyA sites. [From Refseq: http://www.ncbi.nlm.nih.gov/refseq/]. Precursor mRNAAn immature single strand of messenger ribonucleic acid (mRNA) is called a precursor mRNA (pre-mRNA) which is synthesized in the cell nucleus from a DNA template by transcription. Pre-mRNA makes up the majority of heterogeneous nuclear RNA (hnRNA). hnRNA can also include RNA transcripts that do not end up as cytoplasmic mRNA. More details about these proteins can be found at http://atlasgeneticsoncology.org/Genes/GC_HNRNPA1.html and http://www.uniprot.org/uniprot/P09651. The heterogeneous nuclear ribonucleoprotein A1 (hnRNP A1) is sometimes also called “Helix-destabilizing protein”, “Single-strand RNA-binding protein”, or “hnRNP core protein A1” and was identified in the spliceosome C complex, and in a mRNP granule complex, which appears to be at least composed of proteins ACTB, ACTN4, DHX9, ERG, HNRNPA1, HNRNPA2B1, HNRNPAB, HNRNPD, HNRNPL, HNRNPR, HNRNPU, HSPA1, HSPA8, IGF2BP1, ILF2, ILF3, NCBP1, NCL, PABPC1, PABPC4, PABPN1, RPLP0, RPS3, RPS3A, RPS4X, RPS8, RPS9, SYNCRIP, TROVE2, YBX1 and untranslated mRNAs. The protein interacts with SEPT6, with HCV NS5B, and with the 5'-UTR and 3'-UTR of HCV RNA. 5′UTR and 3′UTR are the non-coding regions of HCV RNA, referred herein as 5′ and 3′ untranslated regions that contain important sequence and structural elements critical for HCV translation and RNA replication. Spliceosome
complexes are named according to the metazoan nomenclature. Exon and intron sequences are indicated by boxes and lines, respectively. The stages at which the evolutionarily conserved DExH/D-box RNA ATPases/helicases Prp5, Sub2/UAP56, Prp28, Brr2, Prp2, Prp16, Prp22 and Prp43, or the GTPase Snu114, act to facilitate conformational changes are indicated. (D) Model of interactions occurring during exon definition. (From: Will C L , and Lührmann R Cold Spring Harb Perspect Biol 2011;3:a003707). The DExH/D protein familyThe DExH/D protein family is the largest group of enzymes found in eukaryotic RNA metabolism. DExH/D proteins unwind RNA duplexes in an ATP-dependent fashion. In recent years it has become clear that these DExH/D RNA helicases are also involved in the ATP-dependent remodeling of RNA–protein complexes. DExH/D proteins are essential for all aspects of cellular RNA metabolism and processing, for the replication of many viruses and for DNA replication as well. The DExH/D protein family database contains information about these proteins and makes it available over the WWW (http://www.columbia.edu/~ej67/dbhome.htm). Exon definitionDuring exon definition exons are recognized and defined as units during early assembly by binding of factors to the 3' end of the intron, followed by a search for a downstream 5' splice site. The presence of both a 3' and a 5' splice site in the correct orientation and within 300 nucleotides of one another will allow that stable exon complexes are formed. Concerted recognition of exons may help explain the 300-nucleotide-length maximum of vertebrate internal exons, the mechanism whereby the splicing machinery ignores cryptic sites within introns, the mechanism whereby exon skipping is normally avoided, and the phenotypes of 5' splice site mutations that inhibit splicing of neighboring introns (Roberson et al. 1990). Cytoplasmic mRNP granules at a glance Eukaryotic gene expression is controlled by translation and mRNA degradation which are important in the regulation of these processes. Translation and steps in the major pathway of mRNA decay are in competition with each other. mRNAs that are not engaged in translation can aggregate into cytoplasmic mRNP granules referred to as processing bodies (P-bodies) and stress granules, which are related to mRNP particles that control translation in early development and neurons. The analyses of P-bodies and stress granules suggest a dynamic process. This process is referred to as the mRNA Cycle, wherein mRNPs can move between polysomes, P-bodies and stress granules although the functional roles of mRNP assembly into higher order structures remain poorly understood. (Source: Carolyn J. Decker and Roy Parker; P-Bodies and Stress Granules: Possible Roles in the Control of Translation and mRNA Degradation. Cold Spring Harb Perspect Biol a012286 First published online July 3, 2012). Structure of the Heterogeneous nuclear ribonucleoprotein A1 isoform [Homo sapiens]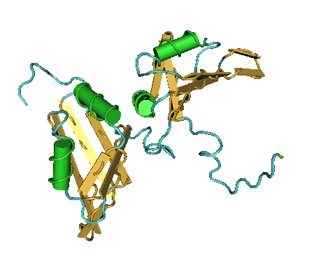 (Source: pdb 2LYV) Chain A, Solution Structure Of The Two Rrm Domains Of Hnrnp A1 (up1) Using Segmental Isotope Labeling PubMed Abstract:“Human hnRNP A1 is a multi-functional protein involved in many aspects of nucleic-acid processing such as alternative splicing, micro-RNA biogenesis, nucleo-cytoplasmic mRNA transport and telomere biogenesis and maintenance. The N-terminal region of hnRNP A1, also named unwinding protein 1 (UP1), is composed of two closely related RNA recognition motifs (RRM), and is followed by a C-terminal glycine rich region. Although crystal structures of UP1 revealed inter-domain interactions between RRM1 and RRM2 in both the free and bound form of UP1, these interactions have never been established in solution. Moreover, the relative orientation of hnRNP A1 RRMs is different in the free and bound crystal structures of UP1, raising the question of the biological significance of this domain movement. In the present study, we have used NMR spectroscopy in combination with segmental isotope labeling techniques to carefully analyze the inter-RRM contacts present in solution and subsequently determine the structure of UP1 in solution. Our data unambiguously demonstrate that hnRNP A1 RRMs interact in solution, and surprisingly, the relative orientation of the two RRMs observed in solution is different from the one found in the crystal structure of free UP1 and rather resembles the one observed in the nucleic-acid bound form of the protein. This strongly supports the idea that the two RRMs of hnRNP A1 have a single defined relative orientation which is the conformation previously observed in the bound form and now observed in solution using NMR. It is likely that the conformation in the crystal structure of the free form is a less stable form induced by crystal contacts. Importantly, the relative orientation of the RRMs in proteins containing multiple-RRMs strongly influences the RNA binding topologies that are practically accessible to these proteins. Indeed, RRM domains are asymmetric binding platforms contacting single-stranded nucleic acids in a single defined orientation. Therefore, the path of the nucleic acid molecule on the multiple RRM domains is strongly dependent on whether the RRMs are interacting with each other.” (Taken from: http://www.rcsb.org/pdb/explore/explore.do?structureId=2LYV). The alignment of 3 hnRNP A1 protein sequences is shown below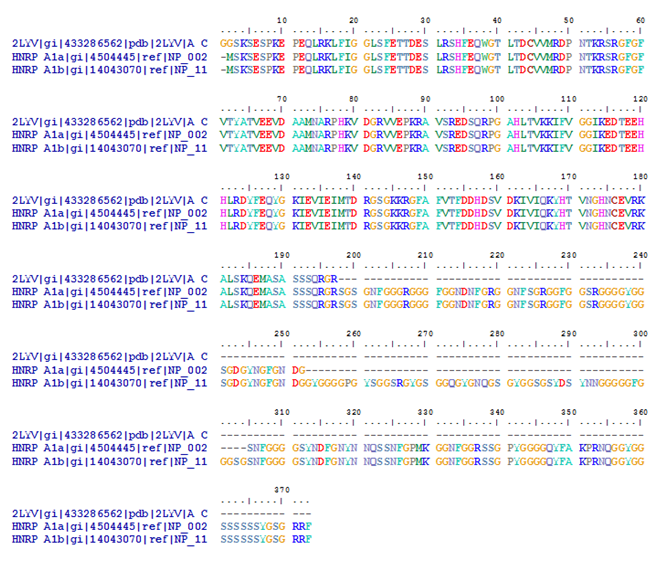 Protein and Peptide Library Information for hnRNP proteinsSolid phase peptide synthesis allows the design and creation of libraries compromising a collection of synthetic peptides. Synthetic libraries have been and can be successfully used for antibody epitope mapping, the determination of the specificity of antibodies, identification of bioactive peptides, development of biological assays, T-cell epitope mapping, vaccine efficacy testing, the screening for ligand-binding activities, screening for antimicrobial peptide activities, peptide-protein interactions, drug discovery, for LC-MS/MS method development and validation as well as for receptor-ligand studies, cellular assays, the study of constrained peptides, modified peptides such as modified histone peptides or the screening for MHC class I and class II peptides, and others. Furthermore peptide libraries provide synthetic, crude or purified peptides that can be customized for the development of screening applications such as epitope mapping, phosphorylation site identifications or the identification of other post-translational modification (PTM) sites, peptide target interaction studies, mid- to high-throughput selected reaction monitoring (SRM) and multiple reaction monitoring (MRM) assays in quantitative mass spectrometry (MS) workflows. Peptide libraries for proteomicsThe study of proteomes, sub-proteomes and protein pathways often requires quantitative MS analysis that depends on the identification and validation of SRM and MRM assays. Peptide libraries offer great convenience and flexibility in the development of multiple applications involving large numbers of peptides including libraries for quantitative MS approaches. The use of peptide libraries greatly reduces the setup time of MS experiments. The SRMAtlas (http://www.mrmatlas.org/) which attempts to map the entire human proteome can be used as a guide for the development of new types of peptide libraries.
Peptides libraries can be derived from the target protein, and, as an option for the use in MS workflows, with either arginine (R) or lysine (K) as the C-terminal amino acid. Synthetic libraries can cover the most commonly used tryptic proteotypic peptides useful for SRM assay development or proteolytic peptides of the whole protein. The maximum peptide length for screening libraries can range from short peptides (5 amino acids) to 20 amino acids. However, for peptide libraries used in MS workflows the standard maximal length is usually 25 amino acids but this length can be increased up to 35 amino acids to ensure that the vast majority of potential proteotypic peptides that are screened for their suitability for a reliable SRM assay are covered.
Example of a peptide library for the hnRNP protein useful for MS workflowsHnrnp A1 protein (up1)pI of Protein: 8.0, Protein MW: 22246, Amino Acid Composition: A10 C2 D13 E18 F10 G17 H8 I8 K16 L8 M4 N4 P6 Q7 R16 S16 T12 V17 W1 Y4
List of Tryptic Peptides
Functional categorization of the proteins for which targeted proteomic assays are available can be found in the MRMAtlas database
The MRMAtlas databaseThe dataset can be found in the MRMAtlas (www.mrmatlas.org or www.srmatlas.org) which is publicly accessible. This database was created as part of the PeptideAtlas project (www.peptideatlas.org) and can be queried via the web-interface for peptides, individual proteins, protein sets, or cellular pathways. These data sets can be used for the design of targeted peptide libraries that can be custom synthesized. If desired, all or selected peptides can be labeled with stable isotopes useful for spiking experiments. Examples of peptide libraries for hnRNP proteins for screening work flowsA: Rrm Domains of HnRNP A1 (up1): Structure ID: PDB: 2LYV_ASequence in FASTA format>gi|433286562|pdb|2LYV|A Chain A, Solution Structure Of The Two Rrm Domains Of Hnrnp A1 (up1) Using Segmental "Isotope Labeling"
Peptide library of 15mers with an overlap of 11 amino acids for HnRNP A1 (up1)
B: Heterogeneous nuclear ribonucleoprotein A1 isoform a [Homo sapiens]Sequence in FASTA format>gi|4504445|ref|NP_002127.1| heterogeneous nuclear ribonucleoprotein A1 isoform a [Homo sapiens]
Peptide library of 15mers with an overlap of 11 amino acids for hnRP A1 a, human
C: Heterogeneous nuclear ribonucleoprotein A1 isoform b [Homo sapiens] {hnRNP A1 b, human}Sequence in FASTA format>gi|14043070|ref|NP_112420.1| heterogeneous nuclear ribonucleoprotein A1 isoform b [Homo sapiens]
Peptide library of 15mers with an overlap of 11 amino acids for hnRNP A1 b, human
ReferencesBarraud P, Allain FH; Solution structure of the two RNA recognition motifs of hnrnp a1 using segmental isotope labeling: how the relative orientation between rrms influences the nucleic acid binding topology.J.Biomol.Nmr (2013) 55 p.119. Carolyn J. Decker and Roy Parker; P-Bodies and Stress Granules: Possible Roles in the Control of Translation and mRNA Degradation. Cold Spring Harb Perspect Biol a012286 First published online July 3, 2012. Frank Desiere, Eric W. Deutsch, Alexey I. Nesvizhskii, Parag Mallick, Nichole King, Jimmy K. Eng, Alan Aderem, Rose Boyle, Erich Brunner, Samuel Donohoe, Nelson Fausto, Ernst Hafen, Lee Hood, Michael G. Katze, Kathleen Kennedy, Floyd Kregenow, Hookeun Lee, Biaoyang Lin, Dan Martin, Jeff Ranish, David J. Rawlings, Lawrence E. Samelson, Yuzuru Shiio, Julian Watts, Bernd Wollscheid, Michael E. Wright, Wei Yan, Lihong Yang, Eugene Yi, Hui Zhang and Ruedi Aebersold Genome Biology 2004, 6:R9 Integration of Peptide Sequences Obtained by High-Throughput Mass Spectrometry with the Human Genome. Eric W Deutsch, Henry Lam & Ruedi Aebersold EMBO reports 9, 5, 429–434 (2008) PeptideAtlas: a resource for target selection for emerging targeted proteomics workflows Domon B, Aebersold R. Science. 2006 Apr 14;312(5771):212-7 Mass spectrometry and protein analysis. Lange V, Malmström JA, Didion J, King NL, Johansson BP, Schäfer J, Rameseder J, Wong CH, Deutsch EW, Brusniak MY, Bühlmann P, Björck L, Domon B, Aebersold R. Mol Cell Proteomics. 2008 Apr 13. Targeted quantitative analysis of Streptococcus pyogenes virulence factors by multiple reaction monitoring. Paola Picotti, Mathieu Clément-Ziza, Henry Lam, David S. Campbell, Alexander Schmidt, Eric W. Deutsch, Hannes Röst, Zhi Sun, Oliver Rinner, Lukas Reiter, Qin Shen, Jacob J. Michaelson, Andreas Frei, Simon Alberti, Ulrike Kusebauch, Bernd Wollscheid, Robert L. Moritz, Andreas Beyer & Ruedi Aebersold Nature. 2013 Feb 14;494(7436):266-70. doi: 10.1038/nature11835. Epub 2013 Jan 20. A complete mass-spectrometric map of the yeast proteome applied to quantitative trait analysis Paola Picotti, Henry Lam, David Campbell, Eric W. Deutsch, Hamid Mirzaei, Jeff Ranish, Bruno Domon and Ruedi Aebersold Nature Methods A database of mass spectrometric assays for the yeast proteome. Nat Methods. 2008 November; 5(11): 913–914. B L Robberson, G J Cote, and S M Berget; Exon definition may facilitate splice site selection in RNAs with multiple exons. Mol Cell Biol. 1990 January; 10(1): 84–94. PMCID: PMC360715 Other resources HNRNPA1: provided by HGNC (http://www.genenames.org/), heterogeneous nuclear ribonucleoprotein A1: provided by HGNC. Primary source: HGNC:5031, See related: Ensembl:ENSG00000135486; HPRD:01242; MIM:164017; Vega:OTTHUMG00000169702; Gene type: protein coding; Organism: Homo sapiens. Lineage: Eukaryota; Metazoa; Chordata; Craniata; Vertebrata; Euteleostomi; Mammalia; Eutheria; Euarchontoglires; Primates; Haplorrhini; Catarrhini; Hominidae; Homo. Also known as: HNRPA1; HNRPA1L3; hnRNP A1; hnRNP-A1 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
↧
BNAs for Duplex and Triplex Formation
BNAs for Duplex and Triplex Formation | |||||||||||||||||||||||||||||||||||||||||||||||||||
2’,4’-BNA (LNA) with a five-membered bridged structure is insufficiently resistant to nucleases, and fully modified 2’,4’-BNA (LNA) oligonucleotides do not have the flexibility required for efficient triplex formation. BNA analogues with increased steric bulk and less conformational restriction were developed. 2’,4’-BNACOC has a seven membered bridged structure and exhibits dramatically improved nuclease resistance. NA-5 = BNACOC: This modification conversely affects duplex stability [i.e., duplexes formed with this nucleic acid analogue are less stable than those formed by 2’,4’-BNA (LNA)]. ENA, with a six-membered bridged structure, has slightly lower duplex-forming ability and significantly higher nuclease resistance than 2’,4’-BNA (LNA). Triplex formation with ENA provided variable results compared to that of 2’,4’-BNA (LNA). 2’,4’-BNANC, has a six-membered bridged structure with a unique structural feature (N-O bond) in the sugar moiety The bridged moiety was designed to have a nitrogen atom, which can:
The nitrogen atom on the bridge can be functionalized by hydrophobic and hydrophilic groups, by steric bulk, or by appropriate functional moieties to:
In addition to the above possibilities, the N-O bond could be cleaved under appropriate reductive conditions to modulate the hybridizing properties of 2’,4’-BNANC.
| 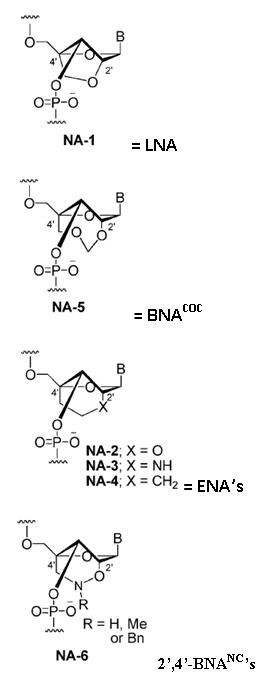 | ||||||||||||||||||||||||||||||||||||||||||||||||||
| |||||||||||||||||||||||||||||||||||||||||||||||||||
| |||||||||||||||||||||||||||||||||||||||||||||||||||
Duplex formation with 2’,4’-BNANC[NH] chimer: | |||||||||||||||||||||||||||||||||||||||||||||||||||
| |||||||||||||||||||||||||||||||||||||||||||||||||||
Duplex formation with 2’,4’-BNA (LNA) chimer: | |||||||||||||||||||||||||||||||||||||||||||||||||||
| |||||||||||||||||||||||||||||||||||||||||||||||||||
| |||||||||||||||||||||||||||||||||||||||||||||||||||
| |||||||||||||||||||||||||||||||||||||||||||||||||||
| |||||||||||||||||||||||||||||||||||||||||||||||||||
Duplex formation with 2’,4’-BNANC[NMe] chimer: | |||||||||||||||||||||||||||||||||||||||||||||||||||
| |||||||||||||||||||||||||||||||||||||||||||||||||||
Duplex formation with 2’,4’-BNA(LNA) chimer: | |||||||||||||||||||||||||||||||||||||||||||||||||||
| |||||||||||||||||||||||||||||||||||||||||||||||||||
| |||||||||||||||||||||||||||||||||||||||||||||||||||
| |||||||||||||||||||||||||||||||||||||||||||||||||||
| |||||||||||||||||||||||||||||||||||||||||||||||||||
a Modified oligonucleotides, 5'-d(GCGTTTTTTGCT)-3'; Target ssRNA strand, 3'-(CGCAAXAAACGA)-5'. b Conditions: 10 mM sodium phosphate buffer (pH 7.2) containing 100 mM NaCl; strand concentration) 4 µM. Conclusion: ΔTmvalues in the Table show that any mismatched base in the target RNA strand resulted in a substantial decrease in the Tm of duplexes formed with 2’,4’-BNANC-modified oligonucleotides. ΔTmvalues of duplexes formed with 2’,4’-BNANC[NH]-modified oligonucleotide having T-U, T-G, and T-C arrangements are -14, -5, -17 °C. These are lower than those of the corresponding natural DNA-RNA duplexes (ΔTm= -12, -3, and -15 °C, respectively). Similar to those exhibited by duplexes formed with the 2’,4’-BNA (LNA) oligonucleotide (ΔTm= -13, -5, and -17 °C, respectively). The mismatch discrimination profiles of 2’,4’-BNANC[NMe] and [NBn] were also similar to that of 2’,4’-BNA (LNA), except in the case of the T-G arrangement for the 2’,4’-BNANC[NMe] derivative, as shown in the Table. Thus, it appears that 2’,4’-BNANC not only exhibits high-affinity RNA selective hybridization, but is also highly selective in recognizing bases. | |||||||||||||||||||||||||||||||||||||||||||||||||||
| |||||||||||||||||||||||||||||||||||||||||||||||||||
| |||||||||||||||||||||||||||||||||||||||||||||||||||
| |||||||||||||||||||||||||||||||||||||||||||||||||||
| |||||||||||||||||||||||||||||||||||||||||||||||||||
| |||||||||||||||||||||||||||||||||||||||||||||||||||


↧
↧
References for BNANC[NH] and [NMe] monomers
References for BNANC[NH] and [NMe] monomers | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
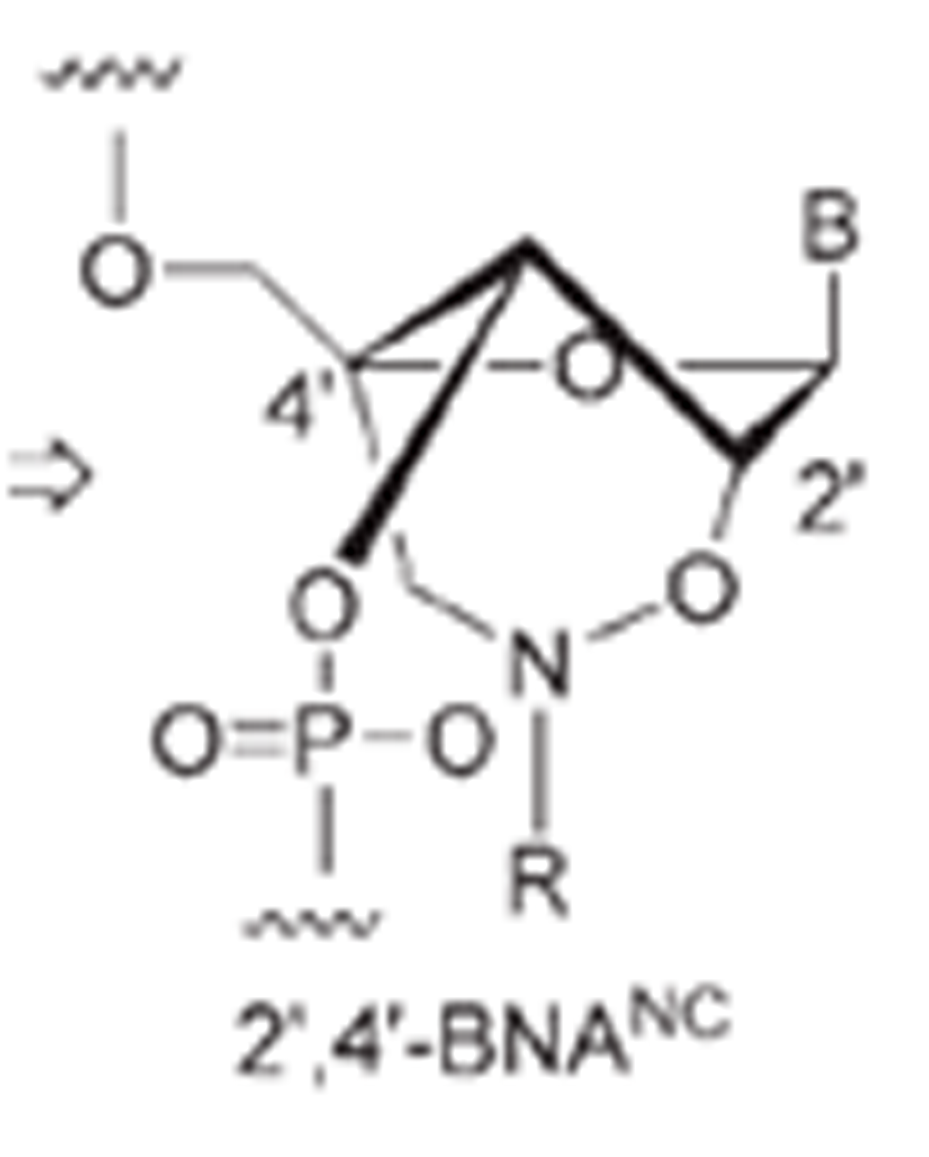
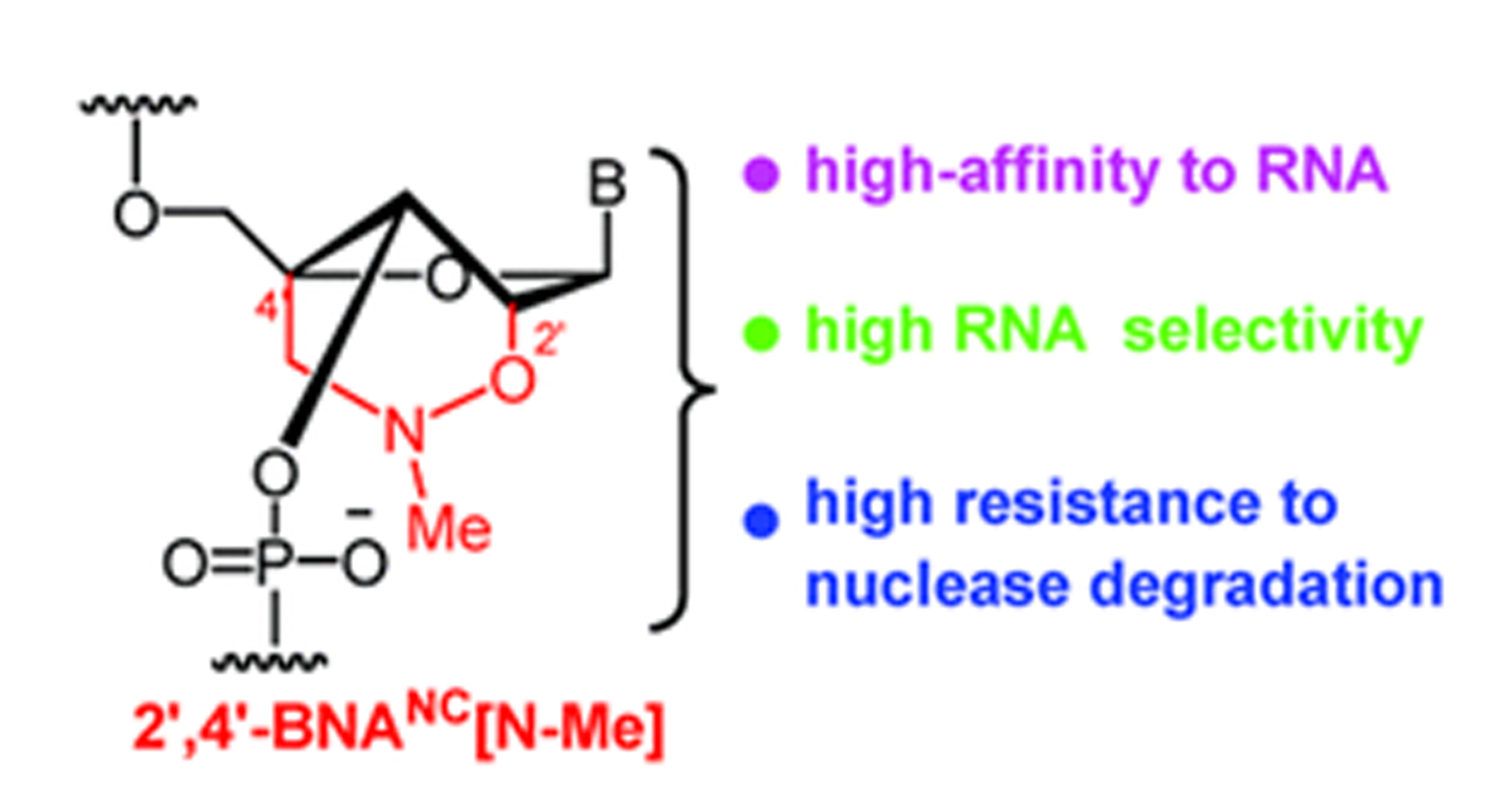
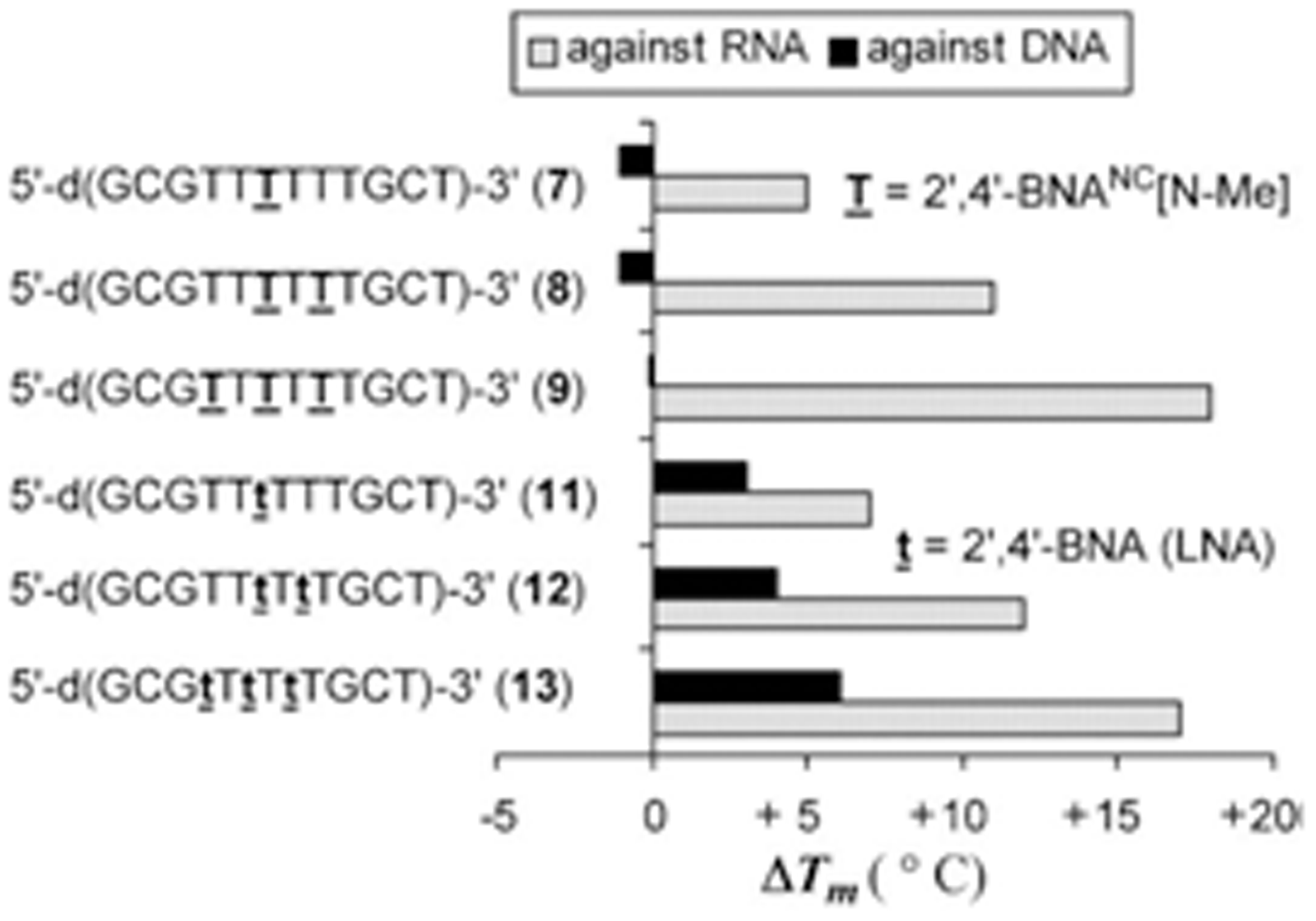
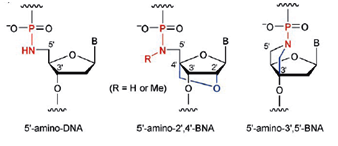
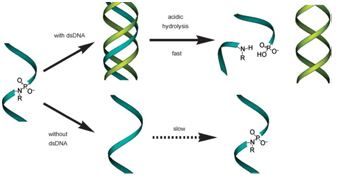
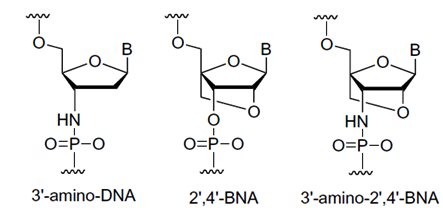

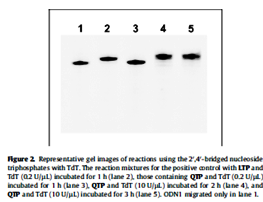
↧
Buffer Used In BNA Experiments
Buffers used in BNA experiments | ||||||||||||||||||||||||||||
| ||||||||||||||||||||||||||||
Example of experimental conditions for FISHTable 1: Different conditions can be tried with FISH technique using BNA/DNA mixmers as probes | ||||||||||||||||||||||||||||
| ||||||||||||||||||||||||||||
The saline-sodium citrate (SSC) buffer is used as a hybridization buffer, to control stringency for washing steps in protocols for Southern blotting, in situ hybridization, DNA Microarray or Northern blotting. 20X SSC may be used to prevent drying of agarose gels during a vacuum transfer. A 20X stock solution consists of 3 M sodium chloride and 300 mM trisodium citrate (adjusted to pH 7.0 with HCl). A 2X SSC buffer consists of 0.3 M sodium chloride and 30.0 mM trisodium citrate (adjusted to pH 7.0 with HCl). Procedure FISH is carried out as described in Table 1. The amount of probe can be varied between 6.4, 10, 13.4 and 20 pmoles. Denaturation of the target DNA and the probe is performed at 75 oC for 5 min either separately using 70% formamide or simultaneously under the coverslip in the presence of hybridization mix containing 50% formamide. In addition, effect of no denaturation is also tested. Two alternative hybridization mixtures can be used: 50% formamide/2xSSC (pH 7.0) / 10% dextran sulphate or 2xSSC (pH 7.0)/10% dextran sulphate. Hybridization times include 30 min, 1, 2, 3 h and overnight. Hybridization temperatures include: 37, 55, 60 and 72 oC. Post washing is either done as for standard FISH, or with 50% formamide/2xSSC at 60 oC, or without formamide. Hybridization signals with biotin labeled BNA/DNA mixmers are visualized indirectly using two layers of fluorescein labeled avidin linked by a biotinylated anti-avidin molecule, which amplifies the signal 8–64 times. The hybridization of Cy3 labeled molecules, however, is visualized directly after a short washing procedure. Slides can be mounted in Vectashield containing 40-60-diamidino-2-phenylindole (DAPI). The whole procedure is carried out in the dark. The signals can be visualized using a Leica DMRB epifluorescence microscope equipped with a SenSys charge-coupled device camera (Photometrics, Tucson, AZ), and IPLAB Spectrum Quips FISH software (Applied Imaging international Ltd, Newcastle, UK) within 2 days after hybridization. In general, twenty metaphases are analyzed after each hybridization experiment. Reference Silahtaroglu AN, Hacihanefioglu S, Guven GS, Cenani A, Wirth J, Tommerup N, Tumer Z. Not para-, not peri-, but centric inversion of chromosome 12. J Med Genet 1998;35(8):682–4. |
↧
What are lncRNAs and lincRNAs
|


↧
solid phase polypeptide synthesis
Solid Phase Polypeptide Synthesis (SPPS) | ||||||||||||||||||||||||||||
The control and regulatory mechanisms for many biological processes are dependent on peptides and proteins derived from α–amino acids. In addition, many modern medicines are now produced from peptides or derivatives of peptides. A few examples are anti-cancer agents, antibiotics or peptide based drugs that control blood pressure. For this reasons α–amino acids and polypeptide chemistry has become a central technology in organic chemistry, biochemistry, biotechnology and medicinal chemistry. The automation of polypeptide synthesis and the many improvements made in peptide synthesis instrumentation in recent years have made synthetic peptides and their derivatives more available to the scientific community and the biological industry as a whole. Synthetic peptides can be used to manufacture epitope-specific antibodies, map antibody epitopes and study enzyme binding sites or to design and synthesize novel peptide- or protein-mimetics or even whole enzymes. To synthesize a peptide a peptide bond between two amino acids will need to be formed. The exact size of peptides is not well defined but it usually refers to flexible chains of up to 30 to 50 amino acids. However, peptides with up to 100 amino acids within the chain can now be synthesized. To synthesize a peptide a peptide bond between two amino acids will need to be formed. The exact size of peptides is not well defined but it usually refers to flexible chains of up to 30 to 50 amino acids. However, peptides with up to 100 amino acids within the chain can now be synthesized. The basic concept in solid-phase peptide synthesis is the step-wise construction of a polypeptide chain attached to an insoluble polymeric support (see Figure 1. for general synthesis scheme). | ||||||||||||||||||||||||||||
| ||||||||||||||||||||||||||||
This approach permits unreacted reagents to be removed by washing without loss of product. Synthesis of the peptide chain proceeds from the carboxyl end of the amino terminus of the polypeptide. The carboxyl moiety of each incoming amino acid is activated by one of several strategies and couples with the α-amino group of the preceding amino acid. The α-amino group of the incoming residue is temporarily blocked in order to prohibit peptide bond formation at this site. The residue is de-blocked at the beginning of the next synthesis cycle. In addition, reactive side chains on the amino acids are modified with appropriate protecting groups. The polypeptide chain is extended by reiteration of the synthesis cycle. Excess reagents are used to drive reactions as close to completion as possible. This generates the maximum possible yield of the final product. After fully assembling the peptide the side-chain protecting groups are removed, and the peptide is cleaved from the solid support, using conditions that inflict minimal damage on labile residues. The product is analyzed to verify the sequence thereafter. The synthetic peptide is usually purified by gel chromatography or HPLC. The blocking group used for blocking the α-amino group determines both the synthesis chemistry employed and the nature of the side-chain protecting groups. The two most commonly used α-amino protecting groups are Fmoc (9-fluorenyl-methoxy-carbonyl) and tBoc (tert.-butyloxycarbonyl). Fmoc side-chain protection is provided by ester, ether and urethane derivatives of tert.-butanol, while the corresponding tBoc protecting groups are ester, ether, and urethane derivatives of benzyl alcohol. The latter are usually modified by the introduction of electron-withdrawing halogens for greater acid-stability. Ether and ester derivatives of cyclopentyl or cyclohexyl alcohol are also employed. The Fmoc protecting group is base-labile. It is usually removed with a dilute base such as piperidine. The side-chain protecting groups are removed by treatment with trifluoroacetic acid (TFA), which also cleaves the bond anchoring the peptide to the support. The tBoc protecting group is removed with a mild acid (usually dilute TFA). Hydrofluoric acid (HF) can be used both to deprotect the amino acid side chains and to cleave the peptide from the resin support. Fmoc is a gentler method than tBoc since the peptide chain is not subjected to acid at each cycle and has become the major method employed in commercial automated polypeptide synthesis. | ||||||||||||||||||||||||||||
| Fmoc Chemistry The α-amino groups are protected by the Fmoc (9-fluorenylmethoxycarbonyl) group, while side-chain protection is provided by ester, ether and urethane derivatives of tert.-butanol (see Figure 2. for details).
Carboxyl group activation of the incoming amino acid utilizes one of the following methods: | ||||||||||||||||||||||||||||
| (1.) BOP/HOBt/NMM: The amino acid is mixed with the BOP (Castro's reagent or (benzotriazol-yloxy) tris (dimethylamino) phosphonium hexa-fluoro-phosphate), HOBt (1-hydro-xybenzotriazole) and suff-icient NMM (N-methyl-morpholine) to ionize 50% of the HOBt. (2.) DIPCDI:DIPCDI (disopropylcarbo-diimide) can be used either with or without HOBt in a solution of DCM:DMF (di-chloromethane and dimethyl-formamide). (3.) Active esters:These are usually pentafluorophenyl esters (OPfp) of the amino acids. The pentafluorophenyl esters of serine and threonine are oils and therefore are employed in the more convenient form of the dihydrooxobenzotriazine ester (ODhbt). tBoc ChemistrytBoc (tert-butyl-oxy-carbonyl) chemistry is widely used in peptide synthesis (see Figure 3.). | ||||||||||||||||||||||||||||
| ||||||||||||||||||||||||||||
Side chain protection is done by ester, ether, and urethane derivatives of benzyl alcohol, and modified by the introduction of electron-withdrawing halogens for greater acid-stability. Ether and ester derivatives of cyclopentyl or cyclohexyl alcohol are also employed. Activation of the incoming amino acid employs DPCDI in a DCM:DMF solution and may also include HOBt. Polypeptide Synthesis Chemistries: A Comparison | ||||||||||||||||||||||||||||
| ||||||||||||||||||||||||||||
| Fmoc-Protected L-Amino Acid Derivatives | ||||||||||||||||||||||||||||
Fmoc amino acids use usually t-butyl based protection for most labile side-chains. The trityl (Trt) protecting group is recommended for Cys. This is removed when the peptide is cleaved with TFA. The acetamidomethyl (Acm) derivative of Cys is unaffected by TFA treatment, but may be rapidly removed by electrophilic reagents such as mercury or iodine. Oxidative deprotection with iodine yields disulfides directly and is a convenient, clean method of forming intramolecular disulfide links. For Arg side-chain protection, both Mtr and the more easily removed Pmc group are widely used. The amidated side-chains of Gln and Asn are protected either with the standard Tmob or with the more recently available Trt. | ||||||||||||||||||||||||||||
| Fmoc-Protected D-Amino Acid Derivatives | ||||||||||||||||||||||||||||
Peptides containing D-amino acids are finding increased use in both research laboratory and pharmaceutical application. Their presence in the peptide allows chain configuration not possible with the L-amino acids alone. Many potent, naturally occurring antibiotics and toxins are cyclic peptides, and often incorporate several D-amino acids to confer a specific conformation as well as make the peptide more resitant to proteolytic attack. |



↧
↧
oligonucleotides containing epigenetic cytosine modifications
Bio-Synthesis provides synthesis of oligonucleotide containing methylated DNA base for epigenetic studies . Epigenetics is the study of changes in heritage control o f gene expression through an identidal DNA sequences , yet maintain different termial phenotypes. This nongenetic cellular memory, which records developmental and environmental cues (and alternative cell states in unicellular organisms), is the basis of epigenetics. The growing interest to fully explain the heritability of complex traits, and to pinpoint the genetic effects in some complex diseases, along with the desire to understand why cells have “deprogramming” into pluripotent/totipotent states, has led epigenetic research into many regulatory systems involving switching a gene from its 'on' to the 'off' state or vise versa in area of DNA methylation, histone modification, nucleosome location, or noncoding RNA. Until 2009, the knowledge about DNA modifications was limited to the introduction of a methyl group at C5 of the cytosine base, which converts 2’-deoxyCytidine (dC) into 5-methyl-2’-deoxyCytidine (mdC).

This methylation occurs predominantly in CpG islands (areas with high occurrence of the CG motif) of promoters. mdC is introduced by specialized DNA methyltransferase (DNMT) enzymes.
In 2009, study showed 1,2 the discovery of 5-hydroxymethyl-2’-deoxyCytidine (hmdC), a novel dC modification in Purkinje neurons and embryonic stem cells. Later, a third report found this modification to be strongly enriched in brain tissues associated with higher cognitive functions.3 This new dC modification is generated by the action of a-ketoglutarate dependent TET enzymes (ten eleven translocation), which oxidizes mdC to hmdC. This finding stimulated discussion about active demethylation pathways that could occur, e.g., via base excision repair (BER), with the help of specialized DNA glycosylases. Alternatively, one could envision a process in which the hydroxymethyl group of hmdC is further oxidized to a formyl or carboxyl functionality followed by elimination of either formic acid or carbon dioxide4,5 .
A number of recent publications provides data that support both pathways. It was discovered that hmdC could be deaminated by activation-induced deaminase (AID) enzymes to provide 5-hydroxymethyl-2’-deoxyUridine (hmdU). This compound was shown to be excised by the SMUG-1 DNA glycosylase6 . After initial failure to detect any further oxidized hmdC derivatives in somatic tissues4, newly developed mass spectrometric technologies, in combination with the available reference compounds, finally enabled researchers to gather strong support for the putative oxidative demethylation pathway. These methods and standards enabled the discovery of 5-formyl-2’-deoxyCytidine (fdC) in differentiating embryonic stem cells.7 Recently, a similar technology also led to the discovery of 5-carboxyl-2’-deoxyCytidine (cdC)8,9, but the amount of fdC and cdC measured differs largely in all three reports.
Research is currently ongoing to unravel the true levels and fate of these further oxidized dC bases in somatic tissues and in different stem cells. Even along the oxidative pathway, base excision processes have been proposed to play a major role with two reports showing that thymidine DNA glycosylase (TDG) accepts both fdC and cdC as substrates.8,10 A possible oxidative demethylation pathway would clearly rely on the existence of a dedicated decarboxylase that is able to convert cdC back into dC. Such an intriguing decarboxylation would enable nature to remove the 5-methyl group in mdC without introducing DNA strand-breaks that accompany any BER based base removal.
Bio-Synthesis has has supported epigenetic resarch by providing the synthesis of oligonucletoides containing all the new cytosine analogues: hmdC, fdC and cdC. These modified base can be incorporate during synthesis at any positions using conventional solid-phase oligonucleotide synthesis chemistry but repalcing the standard cytosine DNA base with mehtylated DNA cytosine base.
- 5-hydroxymethyl-dC [hmdC]
- 5-hydroxymethyl-dC II [hmdCII]
- 5-carboxy-dC [cdC]
- 5-Formyl-dC [fdC]
The first generation hmdC phosphoramidite was fairly very well accepted but requires fairly harsh synthesis conditions. Therefore, a second generation building block (5-Hydroxymethyl-dC II) developed by Carell and co-workers that is compatible with UltraMild deprotection has been introduced.6 5-Formyl-dC and 5-carboxy-dC may find uses in research into DNA damage and repair processes.
References:
- S. Kriaucionis, and N. Heintz, Science, 2009, 324, 929-30.
- M. Tahiliani, et al., Science, 2009, 324, 930-935.
- M. Münzel, et al., Angewandte Chemie-International Edition, 2010, 49, 5375-5377.
- D. Globisch, et al., PLoS One, 2010, 5, e15367.
- S.C. Wu, and Y. Zhang, Nat Rev Mol Cell Biol, 2010, 11, 607-20.
- M. Münzel, D. Globisch, C. Trindler, and T. Carell, Org Lett, 2010, 12, 5671-3.
- M. Münzel, et al., Improved Synthesis and Evaluation of Oligonucleotides Containing 5-Hydroxymethylcytosine, 5-Formylcytosine and 5-Carboxylcytosine. In Chemistry - A European Journal, 2011, in press.
↧
Purification and Quality Control of Long RNA Synthesis by Transcription
Purification of Synthesized Long RNA Transcripts
After long RNA synthesis by transcribing from DNA template, unmodified RNA transcripts from standard DNA synthesis are been purified by phenol-chloroform extraction and ethanol precipitation following with spin column chromatography. For capped RNA syntehsis, non-radioactively labeled RNA or highly specific activity radiolabeled RNA probes, spin column chromatography is the preferred method. When high purity RNA transcript for labeled RAN rpobes for RNase protection assay or foot printing experiments, we recommend gel purificaiton of the transcription product.Bio-Synthesis's Long RNA Synthesis transcription service includes phenol-chloroform extraction and ethanol precipitation. Final product are further purified using spin column chromatography. Gel purification is available with an additional fee.
Characterization of RNA transcription Products
After transcription from template DNA, RNA transcripts concentration are been determined by measuring with ultraviolet light absorbance at 260 nm wavelength. Evaluation of transcript length, integrity and quantity are further analyzed on an appropriate agarose gel or polyacrylamide gel.↧
Heavy Isotope Labeling
Isotopically labeled peptides
Stable-isotope labeled peptides are used in biomarker discovery and validation as well as in drug and metabolite monitoring, peptide signaling experiments, metabolomics and pharmaco kinetics. Bio-Synthesis, Inc is pleased to supply scientists and researchers with custom synthesized stable-isotope peptides. In contrast to standard amino acids, isotopically labeled amino acids are synthesized by substituting 12C, 14N, and H atoms with 13C, 15N, or Deuterium atoms. These amino acids are non-radioactive and have known molecular weights that are higher than standard amino acids. This molecular weight difference makes stable isotope labeled peptides useful tools for quantitative peptide analysis or protein structure and dynamics determination by mass spectrometry (MS) or nuclear magnetic resonance (NMR) spectroscopy, respectively. When used as surrogate markers for proteins of interest, these peptides are added to allow proteins that they represent to be quantified. The best markers/internal standards are peptides that have exact sequences as the biomarker peptides and carry stable isotopic labels. The isotopes change the mass but not the chemical behavior. Isotope ratio of spiked peptides and peptides obtained from protein biomarkers are measured by mass spectrometry and tandem mass spectrometry to provide absolute quantitation of proteins of interest. Stable-isotope labeled peptide sequences are synthesized using the latest Fmoc solid-phase technology and purified by HPLC. All stable-isotope labeled peptide sequences come with certificate of analysis, mass spectrometric analysis and, stringent analytical HPLC to determine the final purity and assure that you are receiving only the highest quality peptides for absolute quantitation.
Stable-isotope labeled peptides are used in biomarker discovery and validation as well as in drug and metabolite monitoring, peptide signaling experiments, metabolomics and pharmaco kinetics. Bio-Synthesis, Inc is pleased to supply scientists and researchers with custom synthesized stable-isotope peptides. In contrast to standard amino acids, isotopically labeled amino acids are synthesized by substituting 12C, 14N, and H atoms with 13C, 15N, or Deuterium atoms. These amino acids are non-radioactive and have known molecular weights that are higher than standard amino acids. This molecular weight difference makes stable isotope labeled peptides useful tools for quantitative peptide analysis or protein structure and dynamics determination by mass spectrometry (MS) or nuclear magnetic resonance (NMR) spectroscopy, respectively. When used as surrogate markers for proteins of interest, these peptides are added to allow proteins that they represent to be quantified. The best markers/internal standards are peptides that have exact sequences as the biomarker peptides and carry stable isotopic labels. The isotopes change the mass but not the chemical behavior. Isotope ratio of spiked peptides and peptides obtained from protein biomarkers are measured by mass spectrometry and tandem mass spectrometry to provide absolute quantitation of proteins of interest. Stable-isotope labeled peptide sequences are synthesized using the latest Fmoc solid-phase technology and purified by HPLC. All stable-isotope labeled peptide sequences come with certificate of analysis, mass spectrometric analysis and, stringent analytical HPLC to determine the final purity and assure that you are receiving only the highest quality peptides for absolute quantitation.
↧
More Pages to Explore .....








